The question that titles this article carries a quiet assumption worth surfacing before we examine a single study: that MOTS-c does modulate glucose homeostasis, and that the only remaining task is to catalog the evidence across conditions. That framing is half right. There is genuine, peer-reviewed evidence that MOTS-c influences glucose handling — but almost all of it sits at the preclinical level, in cultured cells and in mice, and the human data that exist are largely correlational rather than interventional. So the honest version of the question is not “how well does MOTS-c treat dysglycemia?” but “what, exactly, has been shown, in which systems, and how far does it license any claim about people?”
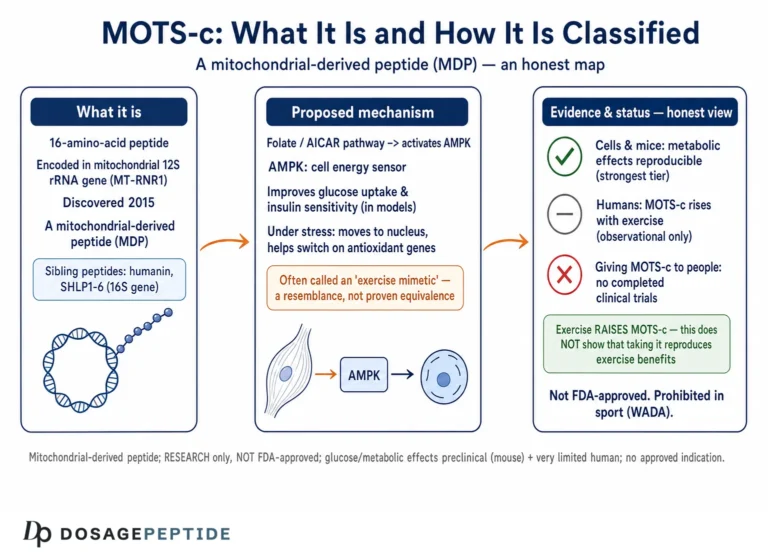
MOTS-c is a 16-amino-acid mitochondrial-derived peptide first described in 2015, when a group at the University of Southern California reported that a short open reading frame hidden within the mitochondrial 12S ribosomal RNA gene encodes a bioactive peptide that improves metabolic homeostasis and reduces insulin resistance in mice.1 That single paper remains the empirical backbone of nearly every glucose-related claim made about the compound. Everything built on top of it — the exercise-mimetic narrative, the aging-and-diabetes hypotheses, the enthusiasm of the research-peptide market — ultimately traces back to a body of rodent work and a handful of human association studies. As of mid-2026, MOTS-c is not approved by the U.S. Food and Drug Administration or any comparable regulator for diabetes, prediabetes, obesity, or any other indication, and no completed Phase 3 program exists.
This piece is written for researchers and scientifically literate readers who want an accurate map of the terrain: what MOTS-c is, the mechanism by which it is proposed to act on glucose metabolism, the preclinical findings that are real and reproducible, the human evidence that is genuinely suggestive but not confirmatory, and the yawning gap between the two. Throughout, the guiding principle is restraint. MOTS-c is an investigational molecule of real scientific interest, not a proven therapy, and nothing here should be read as suggesting it treats, cures, or prevents any disease.
What MOTS-c Is and Why Its Origin Matters
Most of the peptides discussed in metabolic research are either synthetic analogs of gut or pancreatic hormones or fragments of nuclear-encoded proteins. MOTS-c is neither. It belongs to a small and unusual family of mitochondrial-derived peptides (MDPs) — short peptides encoded not in the cell nucleus but within the tiny, circular mitochondrial genome. The name is an acronym: mitochondrial open reading frame of the twelve-S rRNA type-c. The peptide is translated from a sequence embedded inside the 12S rRNA gene, and its full amino-acid sequence is Met-Arg-Trp-Gln-Glu-Met-Gly-Tyr-Ile-Phe-Tyr-Pro-Arg-Lys-Leu-Arg.1
That origin is not a biographical footnote; it is central to why the molecule is interesting for glucose biology. Mitochondria are the organelles where fuel is oxidized and cellular energy is produced, and they sit at the crossroads of glucose and lipid metabolism. The discovery that the mitochondrial genome itself encodes signaling peptides suggested a previously unrecognized layer of communication — a way for mitochondria to broadcast their functional state to the rest of the cell and, via the circulation, to the whole organism.1 MOTS-c and its better-known cousin humanin are the two most studied members of this family. Reviews of the field frame these peptides as retrograde signals that help coordinate the cellular stress response with metabolic demand, which is precisely the kind of biology one would expect to touch insulin action and glucose disposal.2
A useful mental model is to hold three ideas apart. First, MOTS-c is an endogenous molecule: it circulates in human blood and is measurable in tissue, so studying it is partly a matter of understanding normal physiology, not only of evaluating an exogenous drug. Second, it is mitochondrially encoded, which means its expression is tied to mitochondrial abundance and function — a link that becomes important when we consider aging, exercise, and diabetes, all of which alter mitochondrial biology. Third, when researchers or vendors refer to “MOTS-c” as a compound, they mean a synthetic version of that same 16-residue sequence manufactured for laboratory use, which is not the same thing as an approved medicine. Conflating the endogenous signal with the injectable research chemical is one of the most common sources of overstatement in popular writing about the peptide.
Because MOTS-c is discussed alongside a broad catalog of metabolic research peptides, it helps to see where it sits relative to the incretin-based agents that dominate modern glucose therapeutics. The site’s peptide glossary catalogs these mechanistic categories, and the distinction matters: MOTS-c is not an incretin mimetic, not a GLP-1 receptor agonist, and not an insulin analog. It is proposed to act inside the cell, on the machinery of fuel handling itself, rather than on the hormonal signals that tell cells what to do with fuel.
The Proposed Mechanism — Folate Cycle, AMPK, and GLUT4

The mechanistic story for how MOTS-c influences glucose homeostasis is, for a research peptide, unusually well developed at the biochemical level — which is one reason the compound is taken seriously. The original 2015 work proposed a specific and testable pathway rather than a hand-waving “it improves metabolism” claim.1
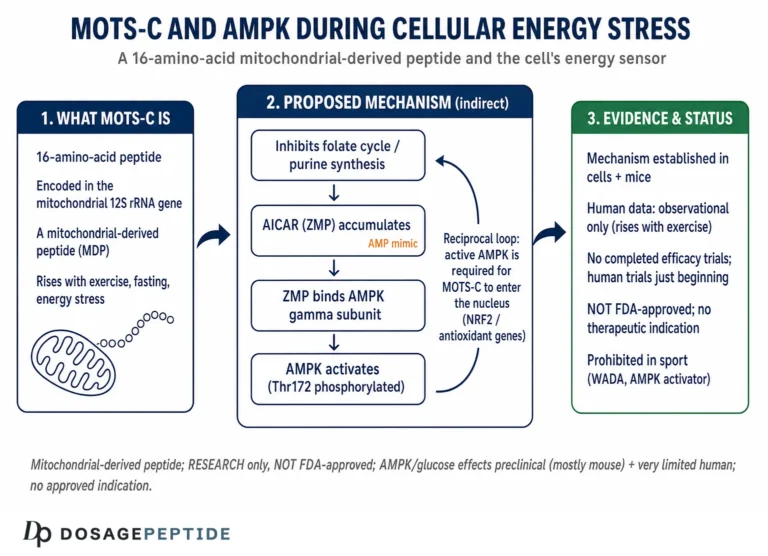
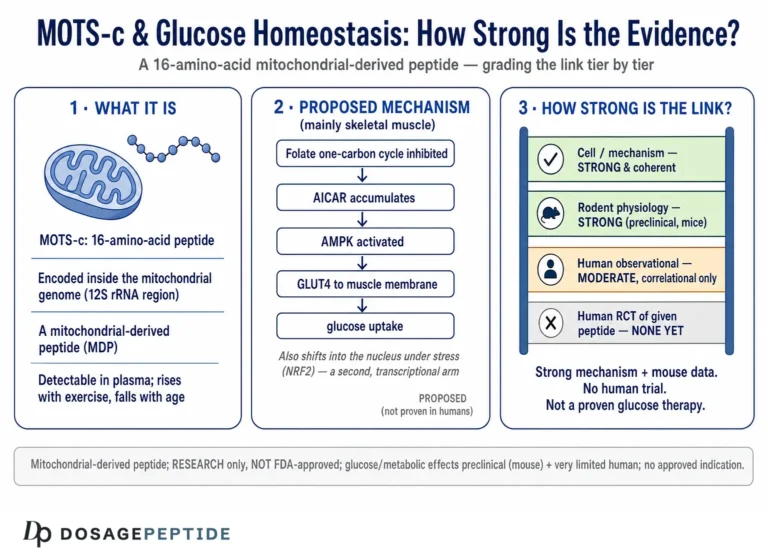
The core idea is that MOTS-c inhibits the folate cycle, the one-carbon metabolic pathway that feeds de novo purine biosynthesis. When that pathway is throttled, an intermediate called AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) accumulates. AICAR is a well-characterized activator of AMP-activated protein kinase (AMPK), the cell’s master energy sensor — indeed, the synthetic compound AICA riboside is used in laboratories precisely because it turns AMPK on. So the proposed sequence is: MOTS-c → folate-cycle inhibition → AICAR accumulation → AMPK activation.1 AMPK, once active, drives a suite of downstream effects that are favorable for glucose disposal: it promotes translocation of the GLUT4 glucose transporter to the cell surface in skeletal muscle, enhances glucose uptake, stimulates fatty-acid oxidation, and shifts the cell toward catabolic, energy-generating metabolism.1
Two features of this mechanism deserve emphasis because they are frequently either overstated or misunderstood. The first is that AMPK activation via AICAR accumulation is, on the evidence, at least partly insulin-independent. In the original mouse work, MOTS-c enhanced glucose uptake in muscle in a manner that did not depend entirely on intact insulin signaling, which is the basis for the recurring claim that the peptide could help glucose disposal even where insulin action is impaired.1 That is a genuinely interesting property, and it distinguishes the proposed mechanism from insulin secretagogues. But “insulin-independent in a mouse muscle preparation” is a long way from “lowers blood glucose reliably in insulin-resistant humans,” and the gap between those statements is exactly where honesty is required.
The second feature is that, unlike the classical route to AMPK activation, MOTS-c is reported to trigger the AICAR-AMPK cascade without first depleting cellular ATP or forcing the cell into an energy-crisis state.2 If accurate, this would make MOTS-c a comparatively “clean” way to engage the AMPK program — pulling the same lever exercise pulls, but through a metabolic rather than an energy-stress trigger. This is the biochemical seed of the “exercise mimetic” framing that dominates lay coverage. It is a reasonable hypothesis grounded in real data, but it is a hypothesis about cellular signaling, not a demonstrated clinical equivalence to exercise, and the distinction is routinely lost.
It is also worth noting that the folate-cycle-to-AICAR-to-AMPK model, while attractive, is not the whole mechanistic story, and subsequent work has complicated it. Later studies have proposed additional or alternative molecular partners — for example, direct binding of MOTS-c to intracellular targets that influence its activity and localization — and reviews of the field are candid that the precise proximal target of MOTS-c remains incompletely defined.2 Some effects may not require the AICAR intermediate at all, and the relative contributions of the cytoplasmic-metabolic route versus the nuclear-transcriptional route to any given glucose outcome have not been fully dissected. For the purposes of the title’s question, the takeaway is that MOTS-c plausibly engages a legitimate insulin-sensitizing pathway, but the mechanism should be described as an active and evolving area of research rather than a settled account, and confident single-sentence mechanistic claims in marketing material tend to flatten a genuinely unfinished picture.
The AMPK-GLUT4 axis is the same downstream machinery targeted, from a different direction, by drugs and lifestyle interventions that improve insulin sensitivity, which is why MOTS-c is often discussed in the same breath as agents studied for fat loss and insulin sensitivity in clinical research. The crucial difference is evidentiary maturity: those incretin agents have completed large randomized trials, whereas MOTS-c’s mechanism, however elegant, has been established mainly in cells and rodents.
Nuclear Translocation and the Stress-Adaptive Gene Program
A second mechanistic layer, described in 2018, deepened the picture and is directly relevant to glucose regulation under stress. Kim and colleagues reported that MOTS-c is not confined to the cytoplasm and mitochondria: under metabolic stress, it translocates to the cell nucleus, where it helps regulate a broad program of stress-adaptive genes.3
The specifics matter. In cell-culture experiments, glucose restriction, serum deprivation, or oxidative stress prompted MOTS-c to move from the mitochondria into the nucleus within about thirty minutes, in a manner dependent on AMPK.3 Once in the nucleus, the peptide was found to associate with stress-responsive transcription factors — notably NRF2 (NFE2L2), a master regulator of antioxidant defenses — and to influence the expression of genes bearing antioxidant response elements (AREs) as well as genes involved in metabolism and glucose handling.3 Strikingly, MOTS-c lacks a canonical nuclear localization signal; its trafficking appears to depend on features of its hydrophobic core rather than the usual address tag, which is part of why the finding was considered novel.
The relevance to glucose homeostasis is twofold. First, it positions MOTS-c as an adaptive responder to precisely the conditions — low glucose, oxidative stress — that a cell encounters during fasting, exercise, or metabolic disease, rather than as a static signal. Second, it links the peptide to the NRF2 antioxidant axis, which connects glucose metabolism to the oxidative-stress biology that pervades diabetic tissue injury. Several of the more recent disease-model studies, particularly in the diabetic heart, lean heavily on this NRF2/ARE mechanism to explain how MOTS-c protects tissue in a hyperglycemic environment.
It is worth being clear-eyed about the level of this evidence. The nuclear-translocation work is elegant molecular cell biology performed largely in cultured cells and mouse tissue. It establishes plausibility and mechanism; it does not establish clinical benefit. A reader who wants to understand the vocabulary of these pathways — AMPK, NRF2, GLUT4, transcription factors — will find them defined in the site’s research glossary, but should keep firmly in mind that mechanistic sophistication and therapeutic proof are different currencies.
The Foundational Rodent Evidence for Glucose Control
Because the 2015 study is the empirical center of gravity for MOTS-c and glucose, it deserves to be described in enough detail that its strengths and its limits are both visible.1
The investigators worked primarily in mice. In diet-induced obesity models, animals fed a high-fat diet (on the order of 60% of calories from fat) develop the rodent analog of insulin resistance: impaired glucose tolerance, reduced insulin sensitivity, and weight gain. When MOTS-c was administered to these mice — by intraperitoneal injection, at doses in the range of 0.5 mg/kg/day over several weeks — the treated animals showed improved glucose tolerance, better insulin sensitivity, and resistance to high-fat-diet-induced weight gain compared with untreated controls.1 Insulin sensitivity was assessed with rigorous methods, including glucose-tolerance testing and hyperinsulinemic-euglycemic clamp studies, the latter being the gold standard for measuring insulin action in vivo. In parallel, the muscle tissue of treated mice showed the expected mechanistic fingerprints: AMPK activation and enhanced GLUT4-mediated glucose uptake.1
Taken on its own terms, this is strong, well-designed preclinical work. It combined a plausible molecular mechanism, appropriate animal models, gold-standard physiological measurements, and a coherent story linking the peptide to a specific glucose-disposal pathway. It is genuinely why MOTS-c earned scientific attention rather than being dismissed as another obscure peptide.
And yet the honest reader must hold several caveats in view simultaneously. The subjects were mice, and rodent metabolic responses translate to humans only unreliably — the history of metabolic drug development is littered with compounds that transformed glucose control in mice and did nothing useful in people. The models were diet-induced obesity and insulin resistance, not established, longstanding type 2 diabetes with beta-cell failure. The administration was by injection at a fixed dose over weeks, not a validated therapeutic regimen. And the endpoints were physiological measures of glucose handling, not the outcomes that matter clinically: sustained HbA1c reduction, prevention of diabetic complications, or hard cardiovascular events. None of this diminishes the quality of the science; it simply locates it accurately as foundational preclinical evidence rather than proof of a human benefit.
What the Human Data Actually Show — and What They Don’t
Here is where discipline is most needed, because the human MOTS-c literature is real, growing, and easily overstated. The critical distinction is between observational studies — which measure how much MOTS-c is present in people with various conditions — and interventional studies, which give people MOTS-c and measure what happens. The overwhelming majority of human MOTS-c research is observational.
The observational signal is reasonably consistent and points in a biologically sensible direction: lower circulating MOTS-c tends to accompany worse metabolic health. In a cross-sectional study of 225 subjects spanning normal glucose tolerance, prediabetes, and type 2 diabetes with good or poor control, serum MOTS-c (alongside the related peptide humanin and the hormone adiponectin) was significantly lower in people with type 2 diabetes than in controls, and levels correlated inversely with HbA1c — those with the worst glycemic control had the lowest MOTS-c.4 Similar reductions have been reported in other insulin-resistant states. In women with polycystic ovary syndrome, a condition tightly linked to insulin resistance and mitochondrial dysfunction, circulating MOTS-c is dynamically regulated by lipid and insulin exposure, and reduced serum and skeletal-muscle MOTS-c has been associated with the mitochondrial dysfunction characteristic of the syndrome.5 Studies in obesity have likewise examined MOTS-c in relation to inflammation, insulin resistance, and endothelial dysfunction, though the obesity findings are notably less consistent than the diabetes signal — some cohorts report no clear difference in circulating levels between obese and lean subjects, a reminder that even the observational picture is not uniform.6
What can we responsibly conclude from this? That MOTS-c is a plausible biomarker of metabolic and mitochondrial health, and that its biology is entangled with glucose regulation in humans. What we cannot conclude is causation or therapeutic benefit. Low MOTS-c in diabetes could be a cause of dysglycemia, a consequence of the mitochondrial dysfunction that accompanies it, or simply a correlate of aging and reduced fitness, since MOTS-c declines with age and rises with exercise. Association studies cannot separate these possibilities. The recurring rhetorical move in vendor and enthusiast writing — “diabetics have low MOTS-c, therefore giving MOTS-c will treat diabetes” — is a textbook correlation-to-causation error. The correlation is real; the therapeutic inference is unproven.
This is the same interpretive caution that applies across metabolic peptide research generally, including the far more mature evidence base for incretin drugs discussed in analyses of whether current evidence supports tirzepatide as a breakthrough therapy for type 2 diabetes. The contrast is instructive: for those agents, the causal chain from mechanism to biomarker to hard clinical outcome has been closed by large randomized trials. For MOTS-c, only the first two links exist, and the third — controlled evidence that administering it improves glucose outcomes in humans — is almost entirely missing.
The K14Q Variant — a Natural Experiment in Glucose Regulation
One of the more compelling human data threads comes not from giving people MOTS-c but from a naturally occurring genetic variant that alters the peptide itself. This is a “natural experiment” of the kind geneticists prize, because it can strengthen causal inference in a way that simple association cannot.
An Asian-specific mitochondrial DNA polymorphism, m.1382A>C, changes a single amino acid in MOTS-c, replacing lysine at position 14 with glutamine — the so-called K14Q variant.7 Researchers led by groups in Japan and the United States examined this variant across large Japanese cohorts. Meta-analysis of tens of thousands of participants indicated that men carrying the C allele had a higher prevalence of type 2 diabetes, an effect most pronounced in sedentary individuals, alongside altered body composition including greater visceral fat.7 Crucially, the group complemented the human genetics with mechanistic experiments: whereas normal MOTS-c improved glucose tolerance and reduced weight in high-fat-fed male mice, the K14Q form did not, indicating that the single-residue change blunts the peptide’s insulin-sensitizing activity.7
This convergence — a variant that both impairs MOTS-c function in the laboratory and associates with more diabetes in the population — is genuinely stronger evidence for a causal role of MOTS-c in human glucose regulation than any correlation of circulating levels. It suggests that MOTS-c biology is not merely a bystander in metabolic disease but participates in it.
Still, three cautions temper the enthusiasm. The gene–environment interaction (the effect being strongest in sedentary men) underscores that MOTS-c operates within a web of lifestyle and physiological factors, not as a solitary switch. The sex-specificity (the association appearing in men but not consistently in women) is unexplained and hints at complexity we do not yet understand. And, most importantly for the therapeutic question, showing that less functional MOTS-c associates with more disease does not demonstrate that administering extra MOTS-c to people with normal or low levels would prevent or treat that disease. The variant strengthens the case that MOTS-c matters; it does not close the case that MOTS-c supplementation works.
Glucose Homeostasis Across Specific Conditions
The title’s phrase — “in various conditions” — invites a survey of the specific disease models in which MOTS-c has been studied for glucose-related effects. The pattern that emerges is consistent: promising, mechanistically coherent findings, almost entirely in animals or cells.
Diet-induced obesity and insulin resistance. This is the best-developed model, anchored by the 2015 work already described, in which MOTS-c improved glucose tolerance and insulin sensitivity and resisted weight gain in high-fat-fed mice.1
The diabetic heart. A cluster of more recent rodent studies has examined MOTS-c in diabetic cardiomyopathy, the heart-muscle injury that accompanies chronic hyperglycemia. In rats made diabetic with a high-fat diet plus low-dose streptozotocin, both aerobic exercise and MOTS-c injection (again around 0.5 mg/kg/day) reduced abnormalities in cardiac structure and function, with the protection attributed to antioxidant and anti-inflammatory effects operating through the NRF2/ARE and NF-κB pathways — the same nuclear program described earlier.8 Related work reported that MOTS-c and exercise restored cardiac function in diabetic rats by engaging NRG1-ErbB signaling.9 These are mechanistically rich preclinical studies, but they address a complication of diabetes in rodents, not glucose control in patients.
Pancreatic islet aging. A 2025 study reported that MOTS-c helps prevent senescence of pancreatic islet cells, potentially delaying diabetes onset in a model system — extending the peptide’s proposed role from peripheral glucose disposal to the insulin-producing beta cells themselves.10 If it holds up, this would be an important addition, because beta-cell failure is central to type 2 diabetes progression. As with the cardiac work, however, it is early-stage model biology.
Aging. Because MOTS-c is mitochondrially encoded and mitochondrial function declines with age, MOTS-c has been positioned as a candidate mediator of age-related metabolic decline, with circulating levels falling as people get older — a trend that overlaps with the age-related deterioration of glucose tolerance.3
The table below summarizes the landscape honestly, foregrounding the evidence level rather than the headline.
| Condition / context | Reported glucose-relevant effect | Highest evidence level to date |
|---|---|---|
| Diet-induced obesity / insulin resistance | Improved glucose tolerance, insulin sensitivity, GLUT4-mediated uptake1 | Mouse (incl. clamp studies) |
| Type 2 diabetes (human) | Lower circulating MOTS-c; inverse correlation with HbA1c4 | Observational / correlational only |
| PCOS / insulin resistance (human) | Reduced serum & muscle MOTS-c; regulated by lipids/insulin5 | Observational / mechanistic |
| K14Q genetic variant (human) | Impaired MOTS-c function associates with more T2D in men7 | Genetic association + mouse function |
| Diabetic cardiomyopathy | Reduced cardiac injury via NRF2/ARE, NRG1-ErbB89 | Rat model |
| Pancreatic islet senescence | Delayed islet-cell senescence, potential diabetes delay10 | Preclinical model |
| Aging | Declining MOTS-c parallels age-related metabolic decline3 | Observational + mouse |
The single most important column is the third. Across every condition, the effect is either measured in animals or inferred from human correlations. Not one row reads “confirmed in an adequately powered randomized controlled trial with clinical endpoints,” because no such trial exists.
The Exercise-Mimetic Framing — Signal and Hype
No discussion of MOTS-c and glucose is complete without addressing the “exercise in a vial” narrative, because it is both the compound’s most seductive selling point and its most abused one.
The framing has a legitimate basis. In 2021, Reynolds and colleagues reported in a major journal that MOTS-c behaves as an exercise-induced, mitochondrially encoded regulator of physical performance and muscle homeostasis.11 In humans, an acute bout of exercise induced a large increase in MOTS-c expression in skeletal muscle and a smaller rise in the circulation, positioning the endogenous peptide as part of the physiological response to exercise itself.11 In mice, MOTS-c treatment enhanced running capacity across young, middle-aged, and old animals, and late-life intermittent treatment improved physical capacity — findings that generated headlines about a “longevity” or “exercise” peptide.11 Because exercise is itself one of the most powerful interventions for improving insulin sensitivity and glucose disposal, a molecule that is induced by exercise and engages the AMPK-GLUT4 pathway naturally invites the hypothesis that it could confer some exercise-like metabolic benefit.12
The honest boundaries of that framing are equally important. First, the human component of even this landmark study was measurement of endogenous MOTS-c in response to exercise, not administration of MOTS-c to improve outcomes; the performance benefits were the mouse findings. Second, exercise produces a vast, coordinated set of adaptations — cardiovascular remodeling, bone loading, neuromuscular coordination, mood and cognitive effects, and system-wide metabolic reprogramming — of which AMPK activation is only one thread. A peptide that pulls the AMPK lever cannot reproduce that orchestra, and describing it as “exercise in a vial” oversells a narrow biochemical overlap as a whole-body equivalence. Third, and most simply: the metabolic benefits of MOTS-c as an exercise mimetic in humans have not been demonstrated in controlled trials. The most defensible statement is that MOTS-c is part of the body’s own exercise-response machinery and shares a key pathway with exercise — not that injecting it substitutes for training or reliably improves human glucose control.
The Only Human Interventional Signal — the CB4211 Analog
If the human MOTS-c literature is overwhelmingly observational, is there any interventional human data? The closest thing comes not from native MOTS-c but from an engineered analog, and the story is instructive precisely because it is modest.
CohBar, a biotechnology company co-founded by one of the MOTS-c discoverers, developed CB4211, described as an improved analog of MOTS-c, and advanced it into a Phase 1a/1b clinical trial for nonalcoholic steatohepatitis (NASH) and obesity. In topline results announced in 2021, the multicenter, randomized, double-blind, placebo-controlled study met its primary endpoint of safety and tolerability, with no serious adverse events reported.13 On exploratory pharmacodynamic measures in the small Phase 1b cohort — roughly twenty obese subjects with fatty liver treated for four weeks — the company reported reductions in liver-enzyme markers (ALT and AST), a decrease in glucose levels, and a trend toward lower body weight versus placebo.13
How much weight should this carry? Some, but carefully bounded. On the positive side, it is the first genuinely interventional human evidence in the MOTS-c space, it was placebo-controlled and double-blind, and it produced a glucose-relevant signal in the expected direction. On the sobering side: it tested an analog, not native MOTS-c; the cohort was tiny; the duration was four weeks; the glucose finding was an exploratory secondary endpoint, not a pre-specified primary outcome, and small early-phase glucose signals frequently evaporate in larger, longer trials; and a Phase 1 study is designed to assess safety, not to establish efficacy. Perhaps most tellingly, the program did not go on to deliver a pivotal efficacy trial that would have converted this promising hint into proof. A single small Phase 1 signal from an analog is a reason for continued scientific interest, not a basis for claiming that MOTS-c treats dysglycemia in people.
How MOTS-c Compares With Established Glucose-Lowering Agents
Placing MOTS-c beside agents with mature evidence is the fastest way to calibrate expectations. The point is not that MOTS-c competes with these drugs — it has never entered a comparable trial — but that the comparison makes the evidentiary distance vivid.
| Agent / class | Primary glucose-relevant mechanism | Highest human evidence | Regulatory status (glucose) |
|---|---|---|---|
| Insulin / analogs | Direct receptor agonism; glucose uptake & suppressed hepatic output | Decades of RCTs; hard outcomes | Approved |
| GLP-1 receptor agonists (e.g., semaglutide) | Glucose-dependent insulin secretion, delayed gastric emptying | Large cardiovascular outcome trials | Approved |
| Dual GIP/GLP-1 agonists (tirzepatide) | Incretin-pathway potentiation; insulin sensitivity gains | Phase 3 program; HbA1c & weight | Approved |
| Metformin | Hepatic gluconeogenesis suppression; AMPK-linked | Decades of RCTs & real-world data | Approved |
| MOTS-c | Folate-cycle inhibition → AICAR → AMPK → GLUT4; insulin-independent uptake | Rodent + human correlational; one small Phase 1 analog signal | Not approved; investigational |
The contrast is stark and should be sat with. Every established glucose-lowering agent has cleared randomized controlled trials measuring HbA1c and, in the leading cases, hard cardiovascular outcomes. MOTS-c has cleared none of these hurdles. Its mechanism is arguably as scientifically interesting as any — the insulin-independent AMPK-GLUT4 route is a real conceptual advantage on paper — but a compelling mechanism is where drug development begins, not where it ends. Readers interested in how a mechanistically related pathway is being explored for glucose and lipid effects can compare the incretin literature on how GLP-1 pathways regulate lipid metabolism, and the diabetes-prevention question being asked of other peptides in work on whether cagrilintide can prevent diabetes in high-risk patients. Those literatures show what a maturing evidence base looks like — and by contrast, how early MOTS-c still is.
Research Models, Methodology, and Handling
Understanding how MOTS-c has been studied clarifies what its data can and cannot support. The research architecture spans three tiers, and knowing which tier a claim rests on is the single most useful analytic habit.
Cellular and molecular work established the mechanism: folate-cycle inhibition, AICAR accumulation, AMPK activation, GLUT4 translocation, and nuclear translocation with NRF2/ARE gene regulation.13 These experiments are the source of mechanistic confidence and are, appropriately, the strongest part of the story at the level of “how might it work.”
Rodent models supplied the physiology: high-fat-diet mice with glucose-tolerance testing and hyperinsulinemic-euglycemic clamps for the metabolic work,1 streptozotocin-plus-high-fat-diet rats for the diabetic-heart work,8 and aged mice for the performance and aging studies.11 Doses cluster around 0.5 mg/kg/day by intraperitoneal injection — a useful fact for interpreting the literature, and a reminder that these are experimental regimens in animals, not human dosing guidance.
Human studies have been overwhelmingly observational: cross-sectional measurements of circulating MOTS-c across metabolic conditions,456 genetic-association analyses of the K14Q variant,7 and measurement of exercise-induced MOTS-c.11 The lone interventional entry is the small Phase 1 analog trial.13 No study has enrolled people with diabetes, administered MOTS-c, and measured HbA1c against placebo over a meaningful duration.
As for handling, MOTS-c is encountered in research settings as a lyophilized (freeze-dried) powder that is reconstituted for laboratory use — a purely educational note, not a usage recommendation, since MOTS-c is not an approved therapeutic. Standard research-peptide practice, described in general terms on the site’s peptide reconstitution guide, applies.
| Parameter | Typical research-context practice |
|---|---|
| Lyophilized storage | Cold and dark; freezing for long-term stability |
| Reconstitution diluent | Bacteriostatic or sterile water, added slowly against the vial wall |
| Mixing | Gentle swirl; never shake (agitation shears peptide bonds) |
| After reconstitution | Refrigerated; used within a limited window |
| Light / heat / freeze-thaw | All degrade peptides; minimize exposure and cycling |
| Purity / sourcing | Research-grade material varies; impurity and mislabeling are real risks |
Meticulous handling changes nothing about the evidence question. A perfectly reconstituted, high-purity vial of MOTS-c is still an investigational compound whose human glucose benefits are unproven. Good technique preserves whatever activity the molecule has; it does not manufacture clinical efficacy.
Limitations and the Evidence Gap
Pulling the threads together, the limitations that bear on the title’s question are substantial and, importantly, they interlock rather than sitting in isolation.
The species gap. The strongest efficacy evidence is in mice and rats. Metabolic physiology translates from rodents to humans notoriously poorly, and the graveyard of failed diabetes and obesity drugs is full of compounds that were spectacular in mice.
The observational-versus-interventional gap. Nearly all human MOTS-c data measure endogenous levels rather than test administration. Low MOTS-c in diabetes is a correlation that cannot, by itself, establish that giving MOTS-c helps. The K14Q variant strengthens causal plausibility, and the CB4211 analog trial provides a small interventional hint, but neither closes the gap to demonstrated therapeutic benefit.
The analog-versus-native gap. The only interventional human signal came from an engineered analog (CB4211), not native MOTS-c, in a tiny, short Phase 1 study on exploratory endpoints — and that program did not proceed to a pivotal efficacy trial.13
The endpoint gap. Even the favorable preclinical work measured physiological glucose handling, not the clinical endpoints regulators require: durable HbA1c reduction, prevention of complications, and cardiovascular safety and benefit. This distinction is not pedantic. Many compounds improve a surrogate measure — fasting glucose, a glucose-tolerance curve, an insulin-sensitivity index — without translating into the outcomes that actually matter to patients over years, and some that move surrogates favorably have later proved harmful on hard endpoints. The regulatory demand for endpoint-driven, adequately powered, long-duration randomized trials exists precisely because surrogate improvements in short studies are an unreliable guide to real-world benefit. For MOTS-c, not a single trial of that kind has been conducted, so the compound sits at the earliest, most uncertain stage of that evidentiary ladder.
Quality and sourcing. Because MOTS-c is not an approved medicine, material sold outside regulated channels varies in purity and provenance, introducing confounders that make even informal observations unreliable.
The intellectually honest position is therefore neither dismissal nor hype. MOTS-c is a real, endogenous mitochondrial peptide with a well-characterized mechanism, reproducible preclinical glucose effects, and a coherent web of human associations suggesting it participates in metabolic health. That is a strong basis for continued investigation. It is not a basis for using MOTS-c to treat or prevent diabetes, and anyone claiming otherwise has skipped the steps — adequately powered, randomized, endpoint-driven human trials — that convert a promising mechanism into a medicine. Readers who want to see how a related mitochondrial and cellular-energy question is examined elsewhere can explore how NAD+ influences cellular repair and longevity in human studies.
Regulatory and Anti-Doping Status
MOTS-c’s regulatory picture is straightforward and frequently misrepresented, so precision matters.
No therapeutic approval, anywhere. MOTS-c is not approved as a drug for diabetes, prediabetes, obesity, NASH, or any other condition by the U.S. Food and Drug Administration, the European Medicines Agency, or any comparable major regulator. It is an investigational molecule. The most advanced clinical effort in the space — the CB4211 analog — reached only Phase 1a/1b before its development did not advance to a pivotal trial.13 There is, correspondingly, no approved indication and no sanctioned therapeutic dose.
Research-chemical status. Synthetic MOTS-c circulating in the research-peptide market is sold for laboratory use only, not as a medicine, and is not manufactured to pharmaceutical standards unless explicitly certified. The distinction between an endogenous human peptide (which everyone possesses) and an injectable research chemical of variable quality (which no regulator has approved for human treatment) cannot be overstated.
Anti-doping prohibition. MOTS-c is prohibited in sport. Anti-doping authorities classify it among substances with metabolic-modulating and performance-relevant properties, and athletes subject to testing should assume that its use constitutes an anti-doping rule violation.14 This is a regulatory hazard entirely independent of the pharmacological questions discussed above.
The regulatory synthesis is simple: MOTS-c is an investigational mitochondrial-derived peptide with no approved medical use, no completed efficacy trial for any glucose indication, and an anti-doping ban in sport. Any legitimate exploration of its metabolic effects belongs in properly authorized preclinical and clinical research under appropriate oversight — not in off-label or informal use.
Frequently Asked Questions
Does MOTS-c lower blood sugar in humans?
There is no adequate randomized controlled trial demonstrating that MOTS-c lowers blood glucose in people. The strongest efficacy evidence is in mice, where MOTS-c improved glucose tolerance and insulin sensitivity in diet-induced obesity models.1 In humans, the data are largely correlational — lower circulating MOTS-c is associated with type 2 diabetes and worse HbA1c — which cannot establish that administering it helps.4 The one interventional human signal came from an analog (CB4211) in a small Phase 1 study on exploratory endpoints.13 MOTS-c is not an approved treatment for diabetes or any other condition.
How is MOTS-c thought to affect glucose metabolism?
The proposed mechanism is that MOTS-c inhibits the folate cycle, causing the intermediate AICAR to accumulate, which activates AMP-activated protein kinase (AMPK). Active AMPK promotes GLUT4 glucose-transporter translocation and glucose uptake in skeletal muscle and enhances fat oxidation.1 Notably, this uptake appears at least partly insulin-independent in preclinical models, and MOTS-c can also translocate to the nucleus under stress to regulate antioxidant and metabolic genes via NRF2.3 This is well-characterized biochemistry, but it has been established mainly in cells and rodents.
Why do people with diabetes have lower MOTS-c?
Circulating MOTS-c is significantly lower in type 2 diabetes and correlates inversely with HbA1c.4 The direction is biologically sensible, since MOTS-c is mitochondrially encoded and diabetes involves mitochondrial dysfunction. But the correlation cannot tell us whether low MOTS-c causes dysglycemia, results from it, or simply reflects shared factors like aging and low fitness. Inferring that supplementation would treat diabetes from this association alone is a correlation-to-causation error.
What does the K14Q genetic variant tell us?
The m.1382A>C variant changes one amino acid in MOTS-c (lysine-14 to glutamine). In large Japanese cohorts, men carrying it — especially sedentary men — had more type 2 diabetes and greater visceral fat, and the variant form failed to improve glucose tolerance in mice that normal MOTS-c helped.7 This convergence of human genetics and mouse function is stronger causal evidence that MOTS-c matters for glucose regulation than circulating-level correlations. It does not, however, prove that giving extra MOTS-c would treat or prevent diabetes.
Is MOTS-c really an “exercise mimetic”?
Partly, and the phrase is easily abused. MOTS-c is induced by exercise in human muscle and circulation and shares the AMPK-GLUT4 pathway that exercise engages, and in mice MOTS-c treatment improved running capacity across ages.11 But exercise produces a vast, coordinated set of adaptations — cardiovascular, skeletal, neuromuscular, cognitive — that a single peptide cannot reproduce, and no controlled human trial has shown that injecting MOTS-c delivers exercise-like metabolic benefit.12 “Part of the body’s exercise-response machinery” is accurate; “a substitute for exercise” is not.
Has MOTS-c been tested in human clinical trials?
Native MOTS-c has not completed efficacy trials in humans. The closest interventional evidence is CB4211, an improved MOTS-c analog developed by CohBar, which completed a Phase 1a/1b study for NASH and obesity. It met its primary safety endpoint and showed exploratory reductions in liver enzymes and glucose plus a trend toward weight loss in about twenty subjects over four weeks — but this was an analog, a tiny cohort, a short duration, and exploratory endpoints, and the program did not advance to a pivotal efficacy trial.13
How does MOTS-c compare with drugs like tirzepatide or metformin?
They are not in the same evidentiary class. Insulin, GLP-1 and dual GIP/GLP-1 agonists, and metformin have all cleared randomized controlled trials measuring HbA1c and, in leading cases, cardiovascular outcomes, and are approved for glucose management. MOTS-c has cleared none of these; its evidence is rodent plus human correlational data and one small Phase 1 analog signal. Its insulin-independent AMPK-GLUT4 mechanism is scientifically interesting, but interesting mechanism is where drug development begins, not where it ends.
Is MOTS-c approved or legal?
MOTS-c is not approved as a drug for any condition by the FDA, EMA, or other major regulators; it is investigational. Synthetic MOTS-c is sold for laboratory research use only, not as a medicine, and material outside regulated channels varies in purity. MOTS-c is also prohibited in sport by anti-doping authorities, so athletes should assume its use is a rule violation.14
How is MOTS-c handled in a research setting?
As a lyophilized powder, it is reconstituted with sterile or bacteriostatic water using gentle technique (swirl, never shake), kept cold and dark, protected from freeze-thaw cycles, and used within a limited window after reconstitution — standard research-peptide practice.2 This is an educational description, not a usage recommendation; handling quality preserves activity but has no bearing on the absence of proven human glucose efficacy.
References
- Lee C, Zeng J, Drew BG, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443-454. PMID: 25738959. https://pubmed.ncbi.nlm.nih.gov/25738959/
- Wan W, Zhang L, Lin Y, Rao X, Wang X, Hua F, Ying J. Mitochondria-derived peptide MOTS-c: effects and mechanisms related to stress, metabolism and aging. J Transl Med. 2023;21(1):36. PMCID: PMC9854231. https://pmc.ncbi.nlm.nih.gov/articles/PMC9854231/
- Kim KH, Son JM, Benayoun BA, Lee C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018;28(3):516-524.e7. PMID: 29983246. https://pubmed.ncbi.nlm.nih.gov/29983246/
- Ramanjaneya M, Bettahi I, Jerobin J, et al. Mitochondrial-derived peptides are down regulated in diabetes subjects. Front Endocrinol (Lausanne). 2019;10:331. PMID: 31214116. https://pubmed.ncbi.nlm.nih.gov/31214116/
- Ramanjaneya M, Jerobin J, Bettahi I, et al. Lipids and insulin regulate mitochondrial-derived peptide (MOTS-c) in PCOS and healthy subjects. Clin Endocrinol (Oxf). 2019;91(2):278-287. PMID: 31066084. https://pubmed.ncbi.nlm.nih.gov/31066084/
- MOTS-c levels in individuals with and without obesity and its association with inflammation, insulin resistance and endothelial dysfunction. PMC (2025). PMCID: PMC12468430. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12468430/
- Zempo H, Kim SJ, Fuku N, et al. A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c. Aging (Albany NY). 2021;13(2):1692-1717. PMID: 33468709. https://pubmed.ncbi.nlm.nih.gov/33468709/
- The role of MOTS-c-mediated antioxidant defense in aerobic exercise alleviating diabetic myocardial injury. Sci Rep. 2023;13:19501. PMCID: PMC10643467. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10643467/
- MOTS-c and exercise restore cardiac function by activating of NRG1-ErbB signaling in diabetic rats. Front Endocrinol (Lausanne). 2022;13:812032. PMCID: PMC8969227. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8969227/
- Kong BS, Lee H, L’Yi S, Hong S, Cho YM. Mitochondrial-encoded peptide MOTS-c prevents pancreatic islet cell senescence to delay diabetes. Exp Mol Med. 2025;57(8):1861-1877. https://www.nature.com/articles/s12276-025-01521-1
- Reynolds JC, Lai RW, Woodhead JST, et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat Commun. 2021;12(1):470. PMID: 33473109. https://pubmed.ncbi.nlm.nih.gov/33473109/
- Zheng Y, Wei Z, Wang T. MOTS-c: a promising mitochondrial-derived peptide for therapeutic exploitation. Front Endocrinol (Lausanne). 2023;14:1120533. PMID: 36761202. https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2023.1120533/full
- CohBar, Inc. CohBar announces positive topline results from the Phase 1a/1b study of CB4211 under development for NASH and obesity. Press release, August 10, 2021. https://www.globenewswire.com/news-release/2021/08/10/2278324/0/en/CohBar-Announces-Positive-Topline-Results-from-the-Phase-1a-1b-Study-of-CB4211-Under-Development-for-NASH-and-Obesity.html
- U.S. Anti-Doping Agency. What is the MOTS-c peptide? (anti-doping status of MOTS-c). https://www.usada.org/spirit-of-sport/what-is-mots-c-peptide/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. MOTS-c is a mitochondrial-derived peptide that is not approved by the FDA, EMA, or any comparable regulator for the treatment, cure, or prevention of type 2 diabetes, prediabetes, obesity, NASH, or any other disease. Its glucose-related effects are supported chiefly by preclinical (cell and rodent) research and human correlational data, with only a single small early-phase interventional signal from an analog; no adequately powered randomized controlled trial has demonstrated a clinical glucose benefit in humans. Nothing here is medical advice or a recommendation for human use. MOTS-c is prohibited in sport by anti-doping authorities. Any legitimate investigation of this compound should occur within properly authorized preclinical or clinical research under appropriate oversight. Readers should consult qualified professionals and applicable regulations before making any decisions.