The question of what MOTS-c “does” during physiological stress is unusually well-posed compared with most peptide questions, because MOTS-c was discovered as a stress-responsive molecule rather than being retrofitted into that role by marketing. It is a 16-amino-acid peptide encoded not in the nuclear genome but inside mitochondrial DNA, within a short open reading frame of the 12S ribosomal RNA gene, and its defining behavior is that it changes its location and activity when a cell’s energy status is threatened.1 So the premise embedded in the title — that MOTS-c has a function in metabolic adaptation to physiological stress — is not a marketing overreach. It is, broadly, what the primary literature describes.
That said, the honest version of the story requires drawing a hard line that popular write-ups routinely blur. There is a large difference between (a) the endogenous, cell-intrinsic role of MOTS-c as a retrograde signal that the body produces in response to exercise, glucose restriction, and oxidative challenge, and (b) the therapeutic use of synthetic MOTS-c injected as a “research peptide” or exercise mimetic. The first is a genuine and increasingly well-characterized piece of cell biology, supported by mechanistic work in cells and mice plus a growing body of human observational data. The second — administering exogenous MOTS-c to healthy people or patients for benefit — rests almost entirely on rodent studies and has never been validated in a controlled human clinical trial. MOTS-c is not approved by the U.S. Food and Drug Administration, the European Medicines Agency, or any comparable regulator for any indication, and there is no established human dose, safety profile, or proven benefit for injected material.
This article therefore does two things at once. It takes the scientific question seriously and lays out, in mechanistic detail, how MOTS-c appears to function within the stress-adaptation machinery of the cell — the nuclear translocation switch, the AMPK and folate–AICAR pathway, the antioxidant-response genes, and the exercise and aging biology that frame all of it. And it keeps a running, explicit accounting of the evidence level behind each claim, because the gap between “this is how the endogenous peptide seems to work in mice and cells” and “therefore injecting it helps humans” is exactly where honest science ends and hype begins. Readers exploring the broader vocabulary of these compounds may find the site’s peptide glossary a useful companion for the terms that recur throughout.
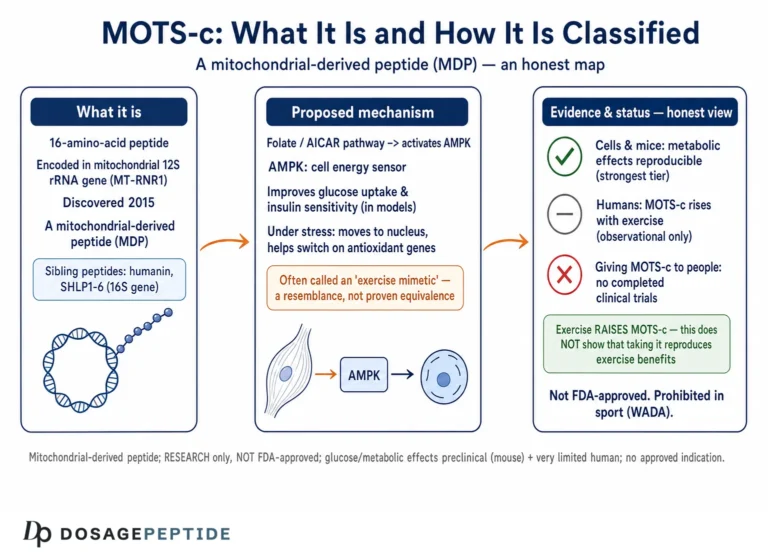
What MOTS-c Is: A Peptide Written in the Mitochondrial Genome
Most of the proteins that operate inside mitochondria are actually encoded by nuclear DNA, translated in the cytoplasm, and imported into the organelle. The mitochondrial genome itself — a small circular molecule of roughly 16,500 base pairs — was long thought to encode only 13 proteins (all subunits of the respiratory chain), plus the transfer RNAs and ribosomal RNAs needed to make them. MOTS-c overturned part of that assumption. In 2015, researchers reported that a short open reading frame hidden within the mitochondrial 12S rRNA gene encodes a functional 16-residue peptide, which they named MOTS-c for “mitochondrial open reading frame of the 12S rRNA type-c.”1 It belongs to an emerging family of so-called mitochondrial-derived peptides (MDPs), the best-known sibling of which is humanin.11
The original characterization established several facts that anchor everything that follows. MOTS-c is detectable not only inside cells but in the circulation, meaning it can behave like a hormone-like signal that travels between tissues. Its principal target organ appears to be skeletal muscle, where it enhances glucose uptake and metabolism. And crucially, its administration to mice prevented diet-induced obesity and both age-dependent and high-fat-diet-induced insulin resistance.1 These effects converged on a single downstream node: activation of AMP-activated protein kinase (AMPK), the cell’s master low-energy sensor.
A subsequent review by the discovering group consolidated the biochemical picture. MOTS-c was shown to target skeletal muscle, enhance glucose disposal while transiently suppressing oxidative respiration, raise cellular NAD+ levels, and activate AMPK — and, importantly for this article’s theme, its circulating and tissue levels decline with age, tracking the emergence of age-related insulin resistance.3 That age-related decline is one of the reasons MOTS-c is so often discussed in the context of stress, exercise, and aging simultaneously: the same molecule sits at the intersection of all three.
It helps to fix the molecular identity clearly before moving to function. MOTS-c is small — sixteen amino acids — which places it well below the size of a folded globular protein and closer to a signaling peptide. Its sequence is encoded on the mitochondrial genome, but a nuance that matters for its stress biology is that the peptide is thought to be translated in the cytoplasm rather than inside the mitochondrial matrix, which is part of what allows it to move freely between compartments, including into the nucleus.2 Holding those three attributes in mind — small, mitochondrially encoded, mobile between compartments — makes the rest of the mechanism intuitive.
The discovery itself is worth understanding because it explains why MOTS-c was almost missed for decades. Conventional gene-annotation tools scan for open reading frames above a size threshold, and short ORFs like the one encoding MOTS-c fell below the radar. Researchers identified it by looking specifically within the 12S rRNA region of the mitochondrial genome for functional short ORFs, then confirming that the predicted peptide was actually made, was present in tissues and plasma, and had reproducible biological activity when synthesized and administered.1 This matters for the honesty of the whole discussion: MOTS-c is not a hypothetical or computationally inferred entity but a physically detected peptide with measured effects. The uncertainty in the field is not about whether MOTS-c exists or is bioactive — those are settled — but about the precise scope, magnitude, and human translatability of its actions. Keeping that boundary clear prevents both the error of dismissing MOTS-c as vaporware and the opposite error of treating its rodent effects as proven human therapy.
The Core Concept: Retrograde Mitonuclear Signaling

To understand MOTS-c’s function in stress adaptation, the single most important idea is retrograde signaling. Ordinarily we think of information flowing from the nucleus outward: nuclear genes are transcribed, proteins are made, and some are shipped to the mitochondria to run metabolism. That is anterograde regulation — the nucleus instructing the organelle. Retrograde signaling is the reverse direction: the mitochondria sending information back to the nucleus about their functional state, so the cell can adjust nuclear gene expression to match metabolic reality.9
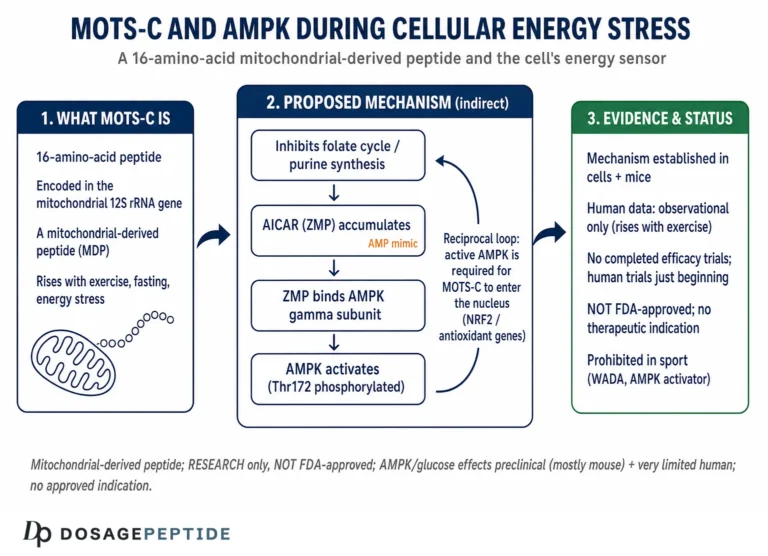
MOTS-c is a striking example of this reverse channel, and arguably the first demonstration that a peptide encoded by mitochondrial DNA can travel to the nucleus and directly influence which nuclear genes are switched on. When researchers showed that MOTS-c translocates from its cytoplasmic/mitochondrial pool into the nucleus under metabolic stress, and there participates in regulating a broad program of nuclear genes, they described it as evidence that mitonuclear communication is genetically integrated in both directions — the mitochondrial genome is not merely a passive power plant but can actively talk back to the nuclear command center.2
This reframes the “function” question. MOTS-c is best understood not as a single-target drug-like molecule but as a stress-coupled coordinator: a signal that reports “energy is scarce / oxidative load is rising” and, in response, helps retune the cell toward conservation, glucose flexibility, and antioxidant defense. In the language of adaptive physiology, it is part of the machinery that lets a cell survive and adjust to a transient insult rather than a molecule that produces one discrete effect. That is precisely why it is discussed under the heading of “metabolic adaptation to physiological stress” rather than under a specific disease.
There is a deeper evolutionary logic that makes this arrangement elegant rather than arbitrary. Mitochondria are descendants of ancient bacteria that were engulfed by an ancestral cell, and over evolutionary time most of their genes migrated to the nucleus — but a small set was retained in the organelle, and those retained genes are overwhelmingly ones whose products must be regulated in tight, local coordination with the respiratory machinery. A peptide encoded in that retained genome, positioned to report the organelle’s stress state directly to the nucleus, is exactly the kind of communication channel one would predict a cell to evolve to keep its two genomes coordinated. MOTS-c can thus be read as a molecular embodiment of mitonuclear co-regulation: the organelle’s way of ensuring that when it is under duress, the nucleus hears about it and responds by adjusting the relevant nuclear gene programs.911
The honest caveat here is about resolution. The existence of MOTS-c nuclear translocation and its association with a stress-responsive gene program are well documented in cell and mouse systems.29 The exact molecular choreography — which import routes it uses, which co-factors it needs, how tissue-specific the program is, and how much of this operates at physiological concentrations in living humans — is still being worked out and is not settled. The mechanism is real and reproducible in its core features; the fine detail is an active research frontier, not a closed textbook chapter. It is also worth flagging that much of the mechanistic work uses concentrations and delivery methods chosen for experimental clarity rather than physiological realism, so the leap from “this pathway operates when we add peptide to cells” to “this is how endogenous MOTS-c behaves at natural levels in a working human tissue” is a real inferential gap that the current literature has not fully closed.
How Physiological Stress Triggers MOTS-c: The Nuclear Translocation Switch
The clearest single experiment behind MOTS-c’s stress role examined what happens under glucose restriction and oxidative stress. Under normal, nutrient-replete conditions, MOTS-c largely stays out of the nucleus. When cells are deprived of glucose or challenged with oxidative stress, MOTS-c rapidly relocates into the nucleus, and this translocation is dependent on AMPK — the same low-energy sensor that MOTS-c helps activate.2 In other words, the trigger for MOTS-c to act as a nuclear regulator is exactly the kind of metabolic emergency the title calls “physiological stress”: falling fuel, rising reactive oxygen species, or the metabolic demand of exercise.
Once inside the nucleus, MOTS-c does not act alone. It associates with stress-responsive transcription factors and regulatory regions, most notably interacting with the pathway governed by nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2), the master regulator of the cellular antioxidant response.2 The net consequence is that MOTS-c helps direct transcription toward genes carrying antioxidant response elements (AREs) and other stress-adaptation programs — effectively converting a mitochondrial distress signal into a coordinated nuclear defensive response.
It is worth laying out the recognized triggers and their reported consequences side by side, because “stress” is doing a lot of work as a word and the specific stimuli are what have actually been studied.
| Physiological stimulus | Reported MOTS-c response | Principal evidence base |
|---|---|---|
| Glucose restriction / caloric scarcity | Nuclear translocation; AMPK-dependent; activation of ARE / stress genes2 | Cell lines, mechanistic |
| Oxidative stress (reactive oxygen species) | Nuclear accumulation; engagement of NRF2/ARE antioxidant program2 | Cell lines, mechanistic |
| Acute exercise | Increased skeletal-muscle and circulating MOTS-c4 | Mouse + limited human |
| Chronic/long-term physical activity | Elevated muscle MOTS-c after weeks of running (rodent)5 | Rodent |
| Metabolic disease (high-fat diet, insulin resistance) | Exogenous MOTS-c restores glucose handling in mice1 | Mouse |
| Aging | Decline in tissue/circulating MOTS-c tracking insulin resistance3 | Mouse + human observational |
Two features of this table deserve emphasis. First, the stimuli that most robustly and mechanistically drive MOTS-c into its nuclear, stress-adaptive mode — glucose restriction and oxidative stress — have been characterized primarily in cellular systems. Second, the stimulus most relevant to everyday human physiology, exercise, is supported by both animal and some human data, which is why exercise gets its own section below. The word “stress” in the title is best read as this cluster of energy- and redox-challenging conditions, not as psychological stress, for which MOTS-c has no established role.
The AMPK and Folate–AICAR Pathway: How MOTS-c Retunes Metabolism
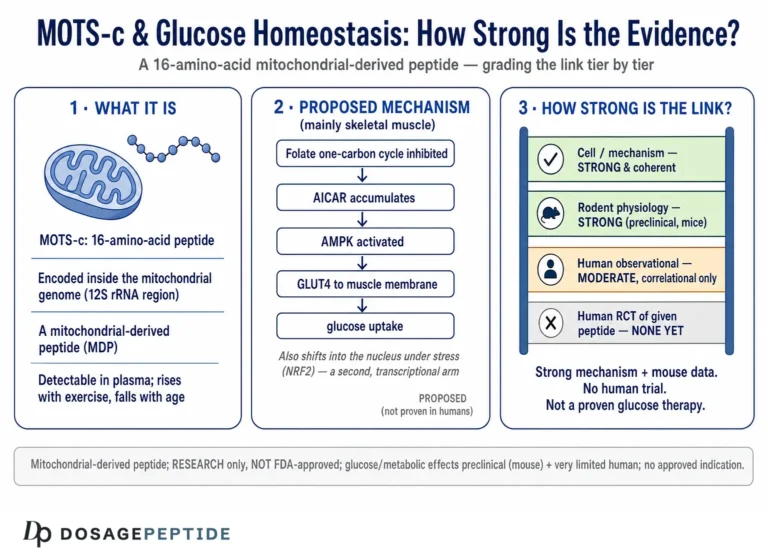
If nuclear translocation is the “where” of MOTS-c’s stress function, the folate–AICAR–AMPK axis is the “how” at the metabolic level. The original mechanistic work proposed that MOTS-c acts on the folate cycle and its tethered de novo purine biosynthesis pathway. By interfering with that pathway, MOTS-c causes the accumulation of an intermediate called AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), which is a well-known endogenous activator of AMPK.13 AICAR is, in fact, the active metabolite of a widely used experimental AMPK-activating compound, so the logic is biochemically grounded rather than speculative.
Why does AMPK activation matter for stress adaptation? AMPK is the cell’s fuel gauge. When the ratio of AMP to ATP rises — the signature of energy stress — AMPK switches on and reorients metabolism toward generating energy and away from consuming it: it promotes glucose uptake, stimulates fatty-acid oxidation, enhances mitochondrial biogenesis through downstream effectors, and restrains anabolic, energy-hungry processes. By feeding into AMPK, MOTS-c effectively amplifies the cell’s own adaptive response to energy shortage, improving metabolic flexibility — the capacity to switch cleanly between fuel sources as conditions change.3
This is also where the insulin-sensitizing observations connect. In mouse models, exogenous MOTS-c improved glucose disposal and protected against insulin resistance induced by aging or a high-fat diet, effects consistent with enhanced AMPK-driven glucose uptake in skeletal muscle.1 That skeletal-muscle glucose-handling angle is why MOTS-c is frequently grouped with metabolic peptides in discussions of insulin sensitivity, even though its mechanism is entirely distinct from incretin-based agents. Readers interested in how a very different class of molecule tackles the same downstream goal of improved insulin sensitivity and fat metabolism can compare the mechanism described here with the clinical incretin literature summarized in how tirzepatide improves fat loss and insulin sensitivity in clinical research — a useful contrast between a mitochondrial retrograde signal and a receptor agonist with large human trial data.
There is an important downstream consequence of AMPK activation that ties the metabolic and antioxidant arms of MOTS-c biology together. AMPK is an upstream activator of PGC-1α, the transcriptional co-activator widely regarded as the master regulator of mitochondrial biogenesis. Through this AMPK–PGC-1α axis, the stress signal that MOTS-c amplifies does not merely help the cell survive an acute energy crisis; it nudges the cell toward building more and better mitochondrial capacity over time — the same adaptive program that endurance training engages. Reviews describe MOTS-c nuclear translocation itself as being driven, at least in part, through an AMPK/PGC-1α-dependent route in response to stress, exercise, and aging, which neatly closes the loop: the peptide both responds to stress and helps reprogram the cell for better future stress tolerance.9 This is why MOTS-c is so often described as an “exercise mimetic” in the popular literature — a label that captures the mechanistic aspiration but overstates the proven reality, since no human intervention has shown that supplying MOTS-c reproduces the benefits of training.
The honest framing of this pathway is that its core steps — folate-cycle interference, AICAR accumulation, AMPK activation, improved glucose handling, and downstream mitochondrial-biogenesis signaling — are supported by consistent mechanistic and rodent data from more than one group, but the quantitative importance of each step in intact humans is not established. AMPK is a hub with many inputs; MOTS-c is one physiological modulator among several (exercise, metformin, and cellular energy state all converge on AMPK), and no human study has isolated MOTS-c’s specific contribution to AMPK signaling in living people. The pathway is a credible mechanism, not a proven human drug target, and the “exercise mimetic” framing should be treated as a hypothesis about what MOTS-c might do rather than a description of what it has been shown to do in people.
MOTS-c and the Antioxidant Response: The Nrf2/ARE Connection
Metabolic stress and oxidative stress are two sides of the same coin. When mitochondria are pushed — by exercise, nutrient scarcity, or dysfunction — they generate more reactive oxygen species, and a cell that cannot manage that oxidative load efficiently will accumulate damage. One of the most interesting aspects of MOTS-c’s stress function is that it does not merely tune energy metabolism; it also appears to arm the cell’s antioxidant defenses.
The mechanistic link runs through NRF2, the transcription factor that governs the expression of a large battery of cytoprotective and antioxidant genes — enzymes involved in glutathione synthesis, detoxification, and redox buffering, all of which contain antioxidant response elements in their promoters. When MOTS-c translocates to the nucleus under oxidative or glucose stress, it engages this NRF2/ARE axis, helping drive the transcription of stress-adaptation genes.2 In this sense MOTS-c functions as an upstream nudge on the master antioxidant switch, coupling the mitochondrial sensing of stress to a nuclear protective response.9
Subsequent work has extended the antioxidant theme into disease-relevant models. Reviews of the field describe MOTS-c involvement in protecting tissues from ischemia–reperfusion injury, inflammatory challenge, and metabolic damage, with the antioxidant/NRF2 program repeatedly implicated as a mediating mechanism.910 These are mechanistically coherent findings, and they reinforce the picture of MOTS-c as a stress-adaptation signal rather than a single-organ hormone.
There is a conceptual elegance worth spelling out here, because it explains why a single peptide would sit at the junction of energy metabolism and redox defense. Mitochondria are simultaneously the cell’s principal energy producers and its principal source of reactive oxygen species — the two functions are physically inseparable, since the same electron-transport chain that makes ATP also leaks the electrons that generate superoxide. A stress that taxes energy production therefore almost always co-occurs with a rise in oxidative load. A signal that responded to only one of these would leave the cell exposed to the other. MOTS-c, by coupling AMPK-driven metabolic retuning to NRF2-driven antioxidant defense, addresses both arms of mitochondrial stress at once, which is precisely the integrated response an efficient adaptive system would deploy. This dual coupling is a large part of why MOTS-c has attracted interest as a candidate node for intervention in conditions where energy failure and oxidative damage travel together — ischemia–reperfusion, metabolic disease, and aspects of aging — even though, again, that interest currently rests on preclinical mechanism rather than human outcome data.910
The essential caveat is the recurring one: this antioxidant biology is characterized in cells and animal models. It is genuinely interesting and reproducible in those systems, and it gives a satisfying mechanistic reason why a metabolic stress signal would also boost antioxidant defense — the two challenges co-occur, so a coordinated response makes evolutionary sense. But “engages the NRF2/ARE program in stressed cells and mice” is not the same claim as “protects human organs from oxidative disease,” and the latter has not been demonstrated in controlled human trials. The antioxidant story is a strong hypothesis with solid preclinical support, not an established clinical fact.
Exercise as the Prototypical Physiological Stress
If one had to choose a single real-world condition that best illustrates MOTS-c’s function in stress adaptation, it would be exercise. Exercise is a controlled, repeated metabolic stress: it transiently depletes energy, raises AMP/ATP ratios, increases reactive oxygen species, and demands rapid fuel switching — precisely the conditions that MOTS-c is built to respond to. And exercise is where the human data, thin as it is, is strongest.
The pivotal study here demonstrated that MOTS-c is exercise-induced. In mice, MOTS-c enhanced physical performance across the lifespan — in young, middle-aged, and old animals — and late-life intermittent MOTS-c treatment increased physical capacity, with old mice roughly doubling their treadmill running capacity and showing improved grip strength and gait.4 Just as importantly, the same study reported that in humans, a single bout of exercise induced MOTS-c expression in skeletal muscle and raised its levels in circulation, establishing that the exercise–MOTS-c link is not purely a rodent phenomenon.4 That human expression finding is one of the load-bearing pieces of evidence that MOTS-c genuinely participates in human exercise physiology.
A separate line of work reinforced the picture from the training side: several weeks of voluntary running increased skeletal-muscle MOTS-c severalfold in rodents, and a single injection of MOTS-c improved acute running time and distance in untrained animals — a demonstration that the peptide can act as a short-term performance modulator, at least in mice.5 Human observational data add nuance and a note of caution against oversimplification: in professional endurance athletes, chronic training was associated with lower resting serum MOTS-c relative to sedentary controls, suggesting the relationship between training status and circulating levels is adaptive and non-linear rather than a simple “more exercise equals more MOTS-c” dial.6 These seemingly contradictory results — acute exercise raises it, chronic elite training may lower resting levels — are exactly what one expects from an adaptive stress signal whose set-point shifts with conditioning.
This is the appropriate place to state the sharpest honesty point in the whole article. The exercise data show that endogenous MOTS-c rises with acute exercise in humans and that exogenous MOTS-c improves performance in mice. They do not show that injecting MOTS-c improves performance, health, or longevity in humans. There is no published randomized controlled trial of administered MOTS-c in people for exercise capacity, metabolic disease, or any other outcome. The leap from “exercise induces this peptide and the peptide helps mice” to “therefore the peptide is a safe, effective exercise mimetic for humans” is precisely the unjustified leap that separates the science from the sales pitch. A parallel example of how energy-metabolism claims for a research peptide are weighed against the actual strength of the evidence appears in the site’s discussion of what studies reveal about KLOW peptides and energy metabolism, which applies the same evidence-grading discipline used throughout this piece. It is also worth stressing that the mouse performance findings, striking as the “doubled running capacity” headline is, were obtained with injected peptide at doses and schedules chosen for the animal model; nothing about those results specifies a dose, a route, a frequency, or a safety margin that would carry over to a human being, and treating the rodent numbers as if they were a human protocol is a category error rather than a conservative extrapolation.
Aging, Insulin Resistance, and the Decline of a Stress Signal
The stress-adaptation function of MOTS-c takes on additional meaning in the context of aging, because aging can be understood in part as a progressive erosion of stress-adaptation capacity. Cells become less able to buffer oxidative load, mitochondrial function declines, and metabolic flexibility narrows. Against that backdrop, one of the most consistently reported observations about MOTS-c is that its levels fall with age.3
The interpretive logic runs as follows. If MOTS-c is a signal the body uses to mount adaptive responses to metabolic and oxidative stress, and if that signal weakens with age, then part of the age-related decline in stress resilience — including the drift toward insulin resistance — might be linked to falling MOTS-c activity. Restoring MOTS-c in aged mice improved metabolic parameters and physical function, which is consistent with this framing.14 This has made MOTS-c a prominent molecule in the broader science of mitochondrial-derived peptides in aging and healthspan, where circulating MDP levels have been correlated with markers such as coronary endothelial function.11
This aging angle also reframes what “metabolic adaptation to physiological stress” means across a lifespan. In a young, metabolically healthy organism, an acute stress — a hard workout, a fast, a transient oxidative challenge — is met by a brisk MOTS-c response that helps the cell adapt and then return to baseline, arguably leaving it slightly more resilient than before (the essence of the training effect). In an aged organism with lower baseline MOTS-c and blunted mitochondrial function, the same stress may provoke a weaker adaptive response, contributing to the reduced stress tolerance and slower recovery that characterize aging. Under this model, MOTS-c is less a discrete “longevity molecule” and more a component of the general adaptive apparatus whose gradual failure is one thread in the larger tapestry of biological aging. It is an attractive and internally consistent hypothesis — but it remains a hypothesis, assembled from association studies and rodent interventions rather than from human trials that manipulated MOTS-c and measured aging outcomes.
It is important, though, to keep the direction of evidence straight. The age-related decline of MOTS-c is an association; whether that decline causes aspects of metabolic aging, or is merely a marker of it, is not resolved. Correlational human data (levels tracking age or metabolic status) and interventional rodent data (restoring MOTS-c helps aged mice) are both suggestive, but neither establishes that raising MOTS-c in aging humans would extend healthspan or reverse insulin resistance. The aging-and-MOTS-c literature is a rich hypothesis-generating body of work, and it is exactly the kind of area where careful human trials are needed and, so far, absent.
Human Genetics: The K14Q Polymorphism as a Natural Experiment
One of the more compelling reasons to take MOTS-c’s human relevance seriously comes not from injected peptide but from genetics. Because MOTS-c is encoded in mitochondrial DNA, naturally occurring variants in its coding region create a kind of natural experiment: populations carrying a variant MOTS-c sequence can be studied for differences in metabolism, fitness, and longevity.
The best-studied variant is a single-nucleotide polymorphism, m.1382A>C, which produces an amino-acid substitution in the MOTS-c peptide (lysine to glutamine at position 14, hence “K14Q”). This variant is essentially specific to Northeast Asian populations. It was first flagged in connection with exceptional longevity, in work examining whether the variant was enriched among long-lived Japanese individuals and might contribute to their metabolic resilience.7 Later and larger analyses complicated the longevity story — the variant’s effect on lifespan is uncertain and may be null — but they simultaneously strengthened the metabolic and physical-performance connections.
Specifically, detailed genetic work found that the K14Q variant is associated with skeletal-muscle fiber composition and muscular performance: carriers showed differences in the proportion of fast-twitch fibers and in measures of muscular strength, with the variant allele appearing at higher frequency in power/sprint athletes than in endurance athletes or controls.8 There is also evidence linking the variant to altered body composition and, in sedentary individuals, to increased type 2 diabetes risk — a gene–environment interaction in which the metabolic downside of the variant emerges specifically when physical activity is low.8
The significance of this genetics for the stress-adaptation question is subtle but real. It provides human evidence, independent of any injected peptide, that the MOTS-c sequence has functional consequences for muscle biology and metabolism — that MOTS-c is not merely an artifact of overexpression experiments but a peptide whose natural variation shapes human physiology. That said, genetic-association data describe correlations across populations, are subject to confounding and population-specific effects, and say nothing about whether administering MOTS-c to a person would be beneficial. The K14Q story validates MOTS-c’s biological importance; it does not validate MOTS-c as a therapy.
What the Evidence Level Actually Is — and Isn’t
Because the central risk with a molecule like MOTS-c is quiet inflation of the evidence, it is worth pausing to grade the major claims explicitly. The pattern that emerges is consistent: strong mechanistic and rodent support for the endogenous stress-adaptation role, genuine but limited human observational support, and essentially no controlled human interventional evidence.
| Claim | Strongest supporting evidence | Evidence level |
|---|---|---|
| MOTS-c is a real mtDNA-encoded peptide present in cells and blood | Original discovery + independent replication13 | Established |
| MOTS-c translocates to the nucleus under metabolic/oxidative stress | Cell-based mechanistic studies2 | Well supported (cell/mouse) |
| MOTS-c activates AMPK via folate–AICAR pathway | Mechanistic + rodent13 | Well supported (preclinical) |
| MOTS-c engages NRF2/ARE antioxidant genes | Cell/mouse mechanistic29 | Supported (preclinical) |
| Exercise induces MOTS-c in human muscle and blood | Human expression data within a mixed study4 | Limited human observational |
| MOTS-c sequence variation affects human metabolism/performance | Genetic association (K14Q)78 | Human observational (association) |
| Injected MOTS-c improves performance / metabolism in mice | Interventional rodent studies45 | Preclinical (rodent) |
| Injected MOTS-c benefits humans (any outcome) | No controlled human trials | Not demonstrated |
The bottom row is the one that vendors and enthusiast sites systematically omit. Everything above it can be discussed with legitimate scientific enthusiasm; the bottom row is where honesty requires a firm stop. MOTS-c is a fascinating piece of endogenous stress biology with a plausible therapeutic future that has not yet been tested in the only setting that could confirm it — adequately controlled human clinical trials. The correct posture toward exogenous MOTS-c is therefore investigational curiosity, not confidence.
This distinction — between a compound’s validated endogenous biology and its unvalidated therapeutic use — recurs across the metabolic-peptide space. It is the same discipline that responsible coverage applies to other research compounds marketed for fat and metabolic effects; the fat-metabolism literature discussed in what clinical trials indicate about the fat-burning potential of AOD-9604 is a useful parallel case of a metabolic peptide whose mechanistic story outran its human efficacy data.
Research Handling and Methodological Context
MOTS-c is encountered in laboratory settings almost exclusively as a lyophilized (freeze-dried) synthetic peptide in a sealed vial, sold for research use. A brief, strictly educational note on how such material is handled in a research context is warranted — with the explicit caveat that describing laboratory handling is not an endorsement of human use, and MOTS-c is not an approved therapeutic for any indication.
The methodological reality behind MOTS-c research is worth understanding because it shapes what the data can and cannot say. The foundational discoveries used a stack of complementary approaches: bioinformatic identification of the short open reading frame within the 12S rRNA gene; synthesis of the peptide for in vitro assays; cell-culture experiments measuring glucose uptake, AMPK phosphorylation, and nuclear localization under glucose restriction; and mouse studies using intraperitoneal injection to test metabolic and performance endpoints.12 The exercise-performance work in mice used defined dosing regimens (for example, daily or intermittent intraperitoneal injection over weeks), which is standard for rodent pharmacology but has no validated human equivalent.4
General research-peptide handling parameters that recur across the literature are summarized below. These are laboratory-practice conventions for preserving peptide integrity, not usage instructions.
| Parameter | Typical research-context practice |
|---|---|
| Lyophilized storage | Cool, dark conditions; freezing favored for long-term stability |
| Reconstitution | Sterile or bacteriostatic water directed against the vial wall; gentle swirl, never shaken |
| After reconstitution | Refrigerated; used within a limited window |
| Light and heat | Minimize exposure; both degrade peptides |
| Freeze–thaw | Repeated cycles degrade peptide; avoid |
| Purity / sourcing | Highly variable in the “research chemical” market; endotoxin and mislabeling are real risks |
The general reconstitution mathematics — how a fixed mass of peptide dissolved in a chosen volume of diluent sets the resulting concentration — is identical across peptides and is walked through in the site’s peptide reconstitution guide; the broader catalog of how these compounds are organized for reference appears on the central dosages index. It bears repeating that meticulous handling changes nothing about the evidence question. A perfectly reconstituted, high-purity vial of MOTS-c is still a compound with no controlled human efficacy data; good technique preserves whatever activity the molecule has but cannot manufacture proof of benefit.
Regulatory and Safety Status
MOTS-c’s regulatory position is straightforward to state and important not to misrepresent. It is not an approved drug. No major regulator — the FDA, the EMA, or comparable authorities — has approved MOTS-c for the treatment, prevention, or cure of any disease, and there is no approved indication in metabolic disease, aging, athletic performance, or anything else. It exists, from a regulatory standpoint, as an investigational substance and a research chemical, not a medicine.
On safety, the honest statement is that the human safety profile of administered MOTS-c is essentially uncharacterized. The favorable-tolerability impressions that circulate online are extrapolated from rodent studies and from the fact that MOTS-c is an endogenous peptide the body already makes — but “the body makes it” is not a safety argument for injecting supraphysiologic amounts of externally sourced material of uncertain purity. There are no published human dose-ranging or safety trials establishing tolerated doses, adverse-event rates, drug interactions, or long-term consequences of exogenous MOTS-c. The absence of reported harm in a setting where almost no controlled human administration has occurred is not evidence of safety; it is an absence of data.
Several additional considerations follow directly from this status:
- Product quality. Because MOTS-c is sold as a research chemical outside regulated pharmaceutical channels, purity, correct sequence, sterility, and endotoxin content are not guaranteed and vary widely by source. These sourcing risks are independent of the molecule’s intrinsic biology.
- Population uncertainty. Rodent metabolic and performance data were generated in defined laboratory animals; extrapolating dose, timing, or effect to humans of varying age, health, and metabolic status has no validated basis.
- Anti-doping considerations. Substances that act as metabolic modulators or purported exercise mimetics can fall within the scope of anti-doping regulation; athletes subject to testing should not assume a research peptide is permitted and should consult current governing-body rules.
- Not a substitute for established interventions. The intervention with by far the strongest evidence for inducing the beneficial stress-adaptation biology described in this article is exercise itself, which reliably raises endogenous MOTS-c and delivers a broad, proven range of metabolic benefits.4
The regulatory synthesis is that MOTS-c occupies the classic position of a promising endogenous signaling molecule at an early, preclinical-to-translational stage: real and interesting biology, an active research literature, reviews openly discussing its therapeutic potential,10 and simultaneously no approval, no validated human dosing, and no proven clinical benefit. Any legitimate exploration of MOTS-c as a therapy belongs in formal, regulated clinical research, not in informal self-administration.
Frequently Asked Questions
What is the function of MOTS-c in metabolic adaptation to physiological stress?
MOTS-c acts as a mitochondria-to-nucleus (retrograde) stress signal. Under energy- or redox-challenging conditions such as glucose restriction, oxidative stress, or exercise, MOTS-c translocates into the nucleus in an AMPK-dependent manner and helps activate stress-adaptation genes, including antioxidant-response (NRF2/ARE) programs, while feeding into the AMPK pathway to improve glucose handling and metabolic flexibility.12 In short, it helps the cell sense metabolic stress and coordinate an adaptive, protective response. This function is well characterized in cells and mice and partly supported by human observational data.
Is MOTS-c produced naturally by the body?
Yes. MOTS-c is an endogenous peptide encoded within a short open reading frame of the mitochondrial 12S rRNA gene. It is detectable in cells and in the circulation, its levels rise with acute exercise and fall with age, and naturally occurring genetic variants in its sequence are associated with differences in human muscle and metabolism.138 This is distinct from injecting synthetic MOTS-c, which is a separate and unproven intervention.
Does taking MOTS-c improve athletic performance in humans?
This has not been demonstrated. In mice, injected MOTS-c improved running capacity and physical function, and in humans a bout of exercise raises endogenous MOTS-c — but there is no published controlled human trial showing that administering MOTS-c improves performance in people.45 The reliable way to raise your own MOTS-c and gain the associated adaptive benefits is exercise itself.
How does MOTS-c relate to AMPK and insulin sensitivity?
MOTS-c is thought to interfere with the folate cycle and de novo purine biosynthesis, causing accumulation of AICAR, an endogenous AMPK activator. AMPK activation promotes glucose uptake and metabolic flexibility, which underlies the improved glucose handling and protection against insulin resistance seen when MOTS-c is given to mice.13 Whether this translates into meaningful insulin-sensitizing effects in humans from administered MOTS-c is untested.
What is the K14Q polymorphism and why does it matter?
K14Q refers to a naturally occurring mitochondrial DNA variant (m.1382A>C) that changes one amino acid in the MOTS-c peptide and is largely specific to Northeast Asian populations. It has been linked to muscle fiber composition, muscular performance, body composition, and — in sedentary individuals — type 2 diabetes risk, providing human genetic evidence that the MOTS-c sequence has real physiological consequences.78 It does not establish that taking MOTS-c is beneficial.
Is MOTS-c approved or proven safe for human use?
No. MOTS-c is not approved by the FDA, EMA, or any comparable regulator for any indication. There are no published human safety or dose-ranging trials of administered MOTS-c, so its human safety profile is essentially uncharacterized, and material sold as a research chemical varies in purity and quality. Absence of reported harm reflects absence of controlled human data, not established safety.
Why do MOTS-c levels change with exercise and age?
As a stress-adaptation signal, MOTS-c rises acutely with exercise, when cells face energy and oxidative demand, and its baseline tends to decline with age as mitochondrial function and stress resilience diminish.34 Interestingly, chronically trained endurance athletes can show lower resting circulating levels, consistent with an adaptive set-point that shifts with conditioning rather than a simple linear response.6
Is MOTS-c the same as humanin or other mitochondrial peptides?
No, though they are related. MOTS-c belongs to the family of mitochondrial-derived peptides (MDPs), which also includes humanin and the small humanin-like peptides. They share a mitochondrial-genome origin and roles in stress and aging biology but differ in sequence, mechanism, and target tissues.11 MOTS-c is distinguished by its skeletal-muscle metabolic focus and its AMPK/folate–AICAR mechanism.
How is MOTS-c studied in the laboratory?
Research on MOTS-c relies on synthesized peptide used in cell-culture assays (measuring glucose uptake, AMPK activation, and nuclear translocation under stress) and in rodent studies using injection at defined regimens to test metabolic and performance endpoints.124 As a lyophilized peptide it follows standard handling practice — cool dark storage, gentle reconstitution, avoidance of freeze–thaw — but good handling has no bearing on the lack of human efficacy data.
References
- Lee C, Zeng J, Drew BG, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443-454. PMID: 25738459. https://pubmed.ncbi.nlm.nih.gov/25738459/
- Kim KH, Son JM, Benayoun BA, Lee C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018;28(3):516-524.e7. PMID: 29983246. https://pubmed.ncbi.nlm.nih.gov/29983246/
- Lee C, Kim KH, Cohen P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med. 2016;100:182-187. PMID: 27216708; PMCID: PMC5116416. https://pubmed.ncbi.nlm.nih.gov/27216708/
- Reynolds JC, Lai RW, Woodhead JST, et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat Commun. 2021;12(1):470. PMID: 33473109; PMCID: PMC7817689. https://www.nature.com/articles/s41467-020-20790-0
- Hyatt JK. MOTS-c increases in skeletal muscle following long-term physical activity and improves acute exercise performance after a single dose. Physiol Rep. 2022;10(13):e15377. PMID: 35808870; PMCID: PMC9270643. https://pmc.ncbi.nlm.nih.gov/articles/PMC9270643/
- Alser M, Ramanjaneya M, Anwardeen NR, et al. The Effect of Chronic Endurance Exercise on Serum Levels of MOTS-c and Humanin in Professional Athletes. Rev Cardiovasc Med. 2022;23(5):181. PMID: 39077591; PMCID: PMC11273660. https://pmc.ncbi.nlm.nih.gov/articles/PMC11273660/
- Fuku N, Pareja-Galeano H, Zempo H, et al. The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell. 2015;14(6):921-923. PMID: 26289118; PMCID: PMC4693465. https://pubmed.ncbi.nlm.nih.gov/26289118/
- Kumagai H, Natsume T, Kim SJ, et al. The MOTS-c K14Q polymorphism in the mtDNA is associated with muscle fiber composition and muscular performance. Biochim Biophys Acta Gen Subj. 2022;1866(2):130048. PMID: 34728329; PMCID: PMC8741734. https://pubmed.ncbi.nlm.nih.gov/34728329/
- Wan W, Zhang L, Lin Y, et al. Mitochondria-derived peptide MOTS-c: effects and mechanisms related to stress, metabolism and aging. J Transl Med. 2023;21(1):36. PMID: 36670507; PMCID: PMC9854231. https://pmc.ncbi.nlm.nih.gov/articles/PMC9854231/
- Zheng Y, Wei Z, Wang T. MOTS-c: A promising mitochondrial-derived peptide for therapeutic exploitation. Front Endocrinol (Lausanne). 2023;14:1120533. PMID: 36761202; PMCID: PMC9905433. https://pmc.ncbi.nlm.nih.gov/articles/PMC9905433/
- Miller B, Kim SJ, Kumagai H, Yen K, Cohen P. Mitochondria-derived peptides in aging and healthspan. J Clin Invest. 2022;132(9):e158449. https://www.jci.org/articles/view/158449
- Sreekumar PG, Kannan R. Going nuclear with stress: MOTS-c and mitonuclear communication. Sci Signal. 2018;11(559):eaav4285. https://www.science.org/doi/10.1126/scisignal.aav4285
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. MOTS-c is a mitochondrial-derived peptide studied as a research compound; it is not approved by the FDA, EMA, or any comparable regulator for the treatment, cure, or prevention of any disease, and there are no controlled human clinical trials establishing the safety or efficacy of administered MOTS-c for metabolic disease, athletic performance, aging, or any other use. The endogenous stress-adaptation biology described here is characterized primarily in cell and animal models, with limited human observational support. Nothing here is medical advice or a recommendation for human use. Any legitimate investigation of this compound should occur within properly authorized preclinical or clinical research under appropriate oversight. Readers should consult qualified professionals and applicable regulations before making any decisions.