The question of which molecular pathways link tesamorelin stimulation to sustained IGF-1 elevation carries a useful tension worth unpacking before we trace any signaling cascade. On one hand, the elevation is unambiguously real: tesamorelin reliably raises circulating insulin-like growth factor-1 (IGF-1), and it does so through a well-characterized chain of events that begins at a single receptor on pituitary cells. On the other hand, the word “sustained” deserves scrutiny. Sustained across what timescale, and in what sense? Sustained the way a continuous infusion holds a level flat, or sustained the way a daily stimulus keeps re-lifting a value that would otherwise fall back? Those are different physiologies, and conflating them is the most common way that writing about this compound drifts from accuracy into marketing.
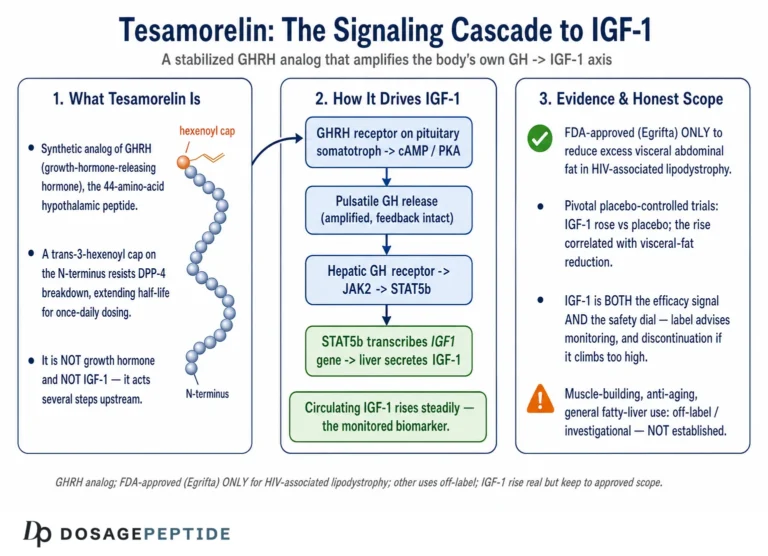
Tesamorelin is a synthetic analog of growth hormone-releasing hormone (GHRH). It is approved by the U.S. Food and Drug Administration under the brand name Egrifta for one indication only: the reduction of excess visceral abdominal fat in adults with HIV-associated lipodystrophy.1 Every other use — anti-aging, body composition in healthy adults, athletic recovery, general “metabolic optimization” — is off-label and rests on far thinner evidence, or none at all. The IGF-1 rise itself was documented in the pivotal HIV trials, where daily tesamorelin produced a substantial increase in IGF-1 relative to placebo.2 So the biochemistry is not in dispute. What this article aims to do is describe honestly how the pathway works, why the resulting IGF-1 elevation is better described as feedback-governed and dosing-dependent than as a fixed new set point, and where the mechanism ends and speculation begins.
This piece is written for researchers and scientifically literate readers who want a mechanistic map rather than a sales pitch. We will walk the pathway from the outside of the somatotroph inward — receptor, G protein, second messenger, transcription factor — then follow growth hormone out of the pituitary and into the liver where most circulating IGF-1 is made, then step back to examine the feedback loops that determine whether the elevation is truly “sustained.” Along the way we will separate what the human trials actually measured from what the mechanism might, in principle, predict. The guiding rule throughout is restraint: an elevated IGF-1 value is a pharmacodynamic marker, not a clinical benefit, and nothing here should be read as evidence that tesamorelin treats, cures, or prevents any condition outside its approved indication.
What Tesamorelin Is: A Stabilized GHRH(1–44) Analog
To understand the pathway, you first have to understand the molecule, because its entire design is aimed at one problem: keeping a fragile hypothalamic signal alive long enough to do its job. Endogenous GHRH is a peptide released from the hypothalamus that travels the short hypophyseal portal route to the anterior pituitary, where it tells somatotroph cells to make and release growth hormone. The trouble is that native GHRH is exquisitely short-lived. The enzyme dipeptidyl peptidase-4 (DPP-4) clips the peptide at its N-terminus within minutes, cleaving the Ala-Asp dipeptide at positions 2–3 and inactivating it almost as fast as it appears.3
Tesamorelin is built to survive that attack. It comprises the full 44-amino-acid sequence of human GHRH(1–44) with one deliberate modification: a trans-3-hexenoyl group is attached to the N-terminal tyrosine.3 That small acyl cap sits precisely where DPP-4 would otherwise grab the peptide, sterically shielding the vulnerable N-terminus from cleavage while leaving the receptor-binding character of the molecule essentially intact. The consequence is a peptide that is recognized by the GHRH receptor as a full agonist but that resists the proteolysis that would otherwise dismantle it. Reported pharmacokinetics reflect this: after subcutaneous injection, tesamorelin reaches peak plasma concentration quickly and carries a plasma elimination half-life on the order of tens of minutes rather than the near-instantaneous turnover of the native hormone.3
It is worth being precise about what this modification does and does not accomplish. It does not turn tesamorelin into a long-acting depot; the peptide is still cleared within an hour or so of a given dose. What it does is buy enough exposure time for a single daily injection to deliver a meaningful, physiologically shaped pulse of GHRH-like signaling to the pituitary. That distinction matters enormously for the “sustained” question. Tesamorelin does not flood the system with a continuous GHRH signal. It provides a stabilized, transient stimulus once per day, and the pituitary responds in its own characteristic pulsatile fashion.1 The sustained IGF-1 elevation seen over weeks of therapy is therefore the aggregate result of repeated daily stimulation, not the product of a drug that holds GHRH signaling continuously high.
This design philosophy places tesamorelin firmly within the family of growth-hormone-axis peptides that work upstream of the pituitary rather than replacing growth hormone itself. It shares conceptual DNA with other GHRH-based analogs; readers exploring that lineage may find the discussion of sermorelin’s role in stimulating natural growth hormone a useful comparison, since sermorelin is a shorter GHRH(1–29) fragment that engages the same receptor by a similar logic but without tesamorelin’s DPP-4-resistant cap. The engineering difference — a stabilized 44-mer versus a native-sequence 29-mer — is exactly the kind of detail that determines potency and duration at the first node of the pathway.
The GHRH Receptor: The First Node in the Pathway
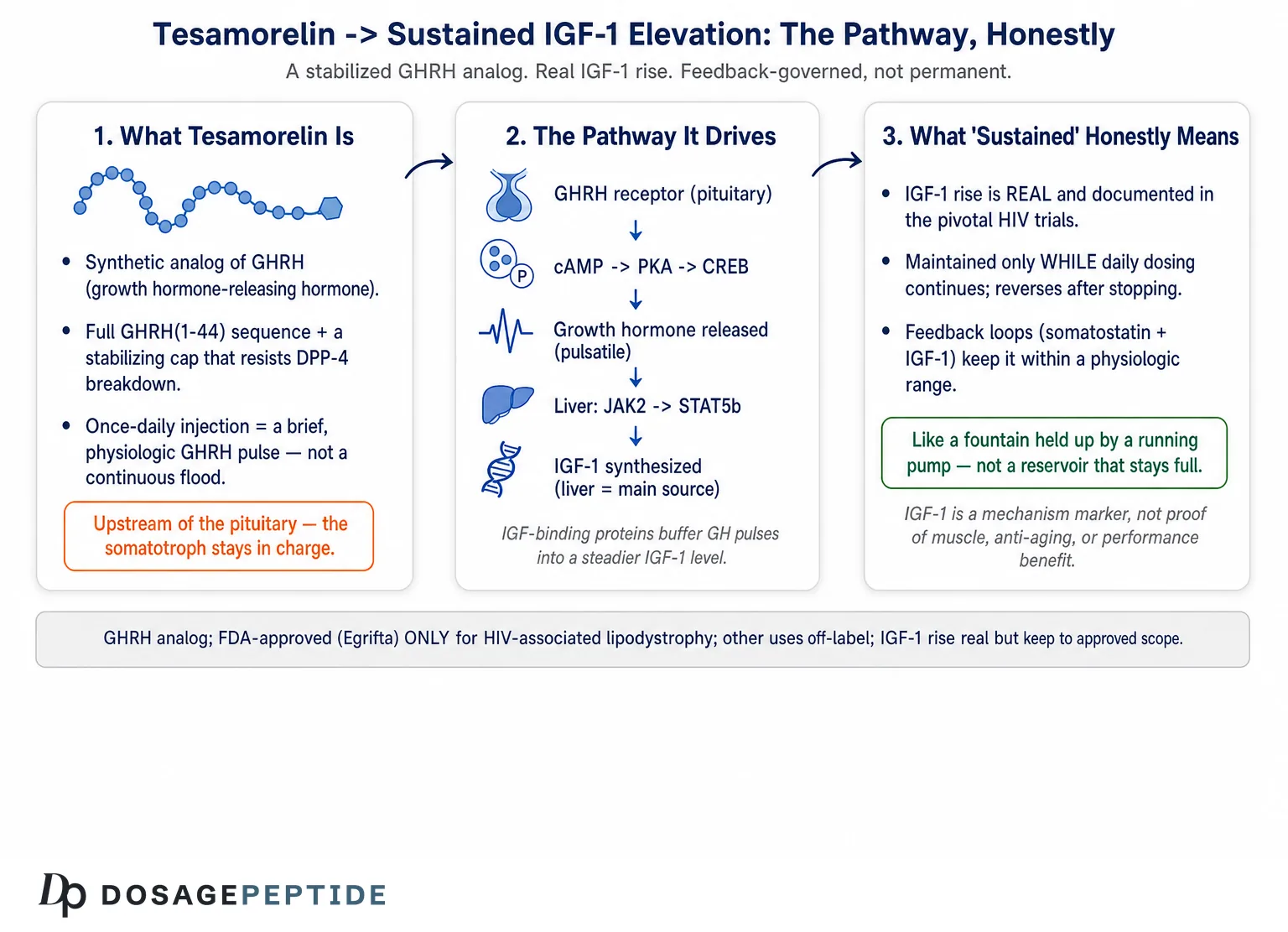
The molecular story of sustained IGF-1 elevation begins at the growth-hormone-releasing hormone receptor (GHRH-R), a class B G-protein-coupled receptor (GPCR) expressed predominantly on the surface of pituitary somatotrophs. This is the target tesamorelin was designed to engage, and everything downstream flows from what happens when the peptide binds.4
When tesamorelin occupies the GHRH-R, the receptor changes conformation and couples preferentially to the stimulatory G protein, activating its Gαs subunit. Gαs in turn switches on adenylyl cyclase, the membrane enzyme that converts ATP into cyclic AMP (cAMP). The rise in intracellular cAMP is the pathway’s first true second-messenger event, and it sets two parallel processes in motion.4
The first process is fast and is responsible for the acute release of stored growth hormone. Elevated cAMP activates protein kinase A (PKA), and the signaling also promotes calcium influx through voltage-gated channels. The resulting rise in intracellular calcium triggers the exocytosis of pre-formed GH-containing secretory granules, so that within minutes of receptor activation the somatotroph discharges a bolus of growth hormone into the circulation.4 This is the acute, secretory arm of the response, and it is why a single subcutaneous dose produces a measurable GH spike.
It is worth pausing on the class B GPCR character of the GHRH receptor, because it explains a subtlety of the pathway that bears on the “sustained” question. Class B (secretin-family) receptors bind their peptide ligands through a two-domain mechanism: a large extracellular N-terminal domain first captures the C-terminal portion of the peptide, after which the peptide’s N-terminus engages the transmembrane bundle to trigger the conformational change that activates Gαs. This is precisely why tesamorelin retains the full GHRH(1–44) sequence rather than being truncated to the minimal signaling core — the length preserves high-affinity binding at the extracellular domain while the stabilized N-terminus drives activation. It is also why the DPP-4-resistant cap does not blunt potency: the modification protects the very residues that must survive to engage the transmembrane pocket. The receptor, in other words, was not merely a target to be hit but a structure whose binding logic dictated the molecule’s design.4
The second process is slower and, for the “sustained” question, arguably more important. PKA phosphorylates the transcription factor CREB (cAMP response element-binding protein). Phosphorylated CREB drives expression of the pituitary-specific transcription factor Pit-1 (POU1F1), which is required for proper transcription of the growth hormone gene in somatotrophs.4 By upregulating Pit-1 and GH gene expression, the cAMP–PKA–CREB cascade does more than release existing hormone — it replenishes the cellular stores of growth hormone so that the somatotroph is prepared to respond to the next stimulus. In other words, repeated GHRH-R activation not only empties the granule pool but also refills it, and even supports somatotroph proliferation and hypertrophy under sustained stimulation.4
This dual output — acute secretion plus transcriptional replenishment — is the mechanistic foundation for why daily tesamorelin can raise IGF-1 for weeks rather than exhausting the pituitary after a few doses. Each day’s injection triggers release; the accompanying transcriptional program restocks the cell. It is a fundamentally different arrangement from simply injecting growth hormone, because the somatotroph remains the gatekeeper and continues to manufacture and package its own product. The distinction between engaging the receptor upstream versus supplying the hormone directly is a recurring theme across the growth-hormone secretagogue field; the mechanistic contrast with receptor-selective agents is explored in the analysis of ipamorelin’s precision in targeting the GHSR-1a receptor, which acts through an entirely separate receptor (the ghrelin receptor) to release GH by a complementary route.
From Somatotroph to Hepatocyte: How GH Becomes IGF-1
Raising growth hormone is only the middle of the pathway. The endpoint named in the title — IGF-1 elevation — happens mostly in the liver, one biochemical step removed from the pituitary. Understanding that step is essential, because it is where the “sustained” character of the response is actually generated and buffered.
Once tesamorelin-stimulated growth hormone enters the bloodstream, it circulates to peripheral tissues and binds the growth hormone receptor (GHR), which is expressed densely on hepatocytes. GH binding dimerizes the receptor and activates the receptor-associated tyrosine kinase JAK2, which phosphorylates and recruits STAT5 (predominantly STAT5b). Phosphorylated STAT5b translocates to the nucleus and drives transcription of the IGF-1 gene, among many other GH-responsive targets.5 The liver is the dominant source of the IGF-1 that circulates systemically, so hepatic GHR–JAK2–STAT5b signaling is the proximate molecular engine behind the IGF-1 rise that tesamorelin produces.5
Several features of this hepatic step shape the kinetics of the IGF-1 response and explain why it looks “sustained” even though the driving GH pulses are brief. IGF-1 has a far longer circulating half-life than growth hormone itself, in large part because the vast majority of it does not float free but is carried in ternary complexes with IGF-binding protein-3 (IGFBP-3) and the acid-labile subunit (ALS), both of which are themselves GH-dependent. This binding-protein reservoir smooths the sharp, pulsatile GH signal into a comparatively stable IGF-1 concentration. The pituitary speaks in pulses; the liver and its binding proteins translate those pulses into a steadier tone. That translation is precisely why a once-daily peptide that produces transient GH spikes can nonetheless yield an IGF-1 value that stays elevated across the dosing interval and, with continued therapy, across weeks.
It also means that the IGF-1 elevation reported in clinical studies is not a direct readout of any single GH pulse but an integrated, buffered summary of the pathway’s recent activity. When the pivotal HIV trials reported mean IGF-1 increases of roughly 100 ng/mL or more relative to placebo, that number reflected the steady-state balance struck between daily GHRH-R stimulation and the whole downstream apparatus of GH secretion, hepatic transduction, and binding-protein carriage.26 The elevation is real, it is measurable, and it is mechanistically coherent — but it is a system-level equilibrium, not a switch flipped to a new position.
There is a further layer worth naming, because it distinguishes a GHRH analog from cruder ways of raising IGF-1. Growth hormone does not act on the liver in a vacuum; the amplitude and, critically, the pulsatile pattern of GH exposure influence how hepatocytes respond, including the sex-dependent programming of GH-responsive hepatic genes governed by STAT5b. A pulsatile GH signal and a continuous one are not biologically interchangeable at the level of hepatic gene expression, even if they produced the same average GH concentration. Because tesamorelin preserves the pituitary’s native pulsatile output rather than imposing a flat exogenous level, the IGF-1 it generates is produced under a physiologically patterned GH signal — a qualitative feature of the pathway that direct hormone administration cannot reproduce.5 Whether that pattern-preservation confers any clinical advantage is not established, but mechanistically it is a real difference, and it is part of why the compound’s IGF-1 elevation is described as physiologic rather than merely quantitative.
Why “Sustained” Is the Right Word — and Where It Isn’t
Now we can address the title’s premise head-on. In what sense is the IGF-1 elevation sustained, and in what sense is that word potentially misleading?
It is fair to call the elevation sustained in two specific ways. First, within a dosing interval: because of the binding-protein reservoir described above, IGF-1 does not spike and crash with each injection the way growth hormone does; it holds a relatively even elevated concentration between daily doses. Second, over the course of continued therapy: the pooled analysis of the phase 3 HIV trials, together with their safety-extension data, showed that the IGF-1 increase and the visceral-fat effect were maintained across many months of continuous daily dosing without the response fading away.6 The pituitary did not become refractory; the pathway kept working. In that longitudinal sense, “sustained” is accurate and evidence-based.
But there are two important senses in which “sustained” must be qualified, and honest mechanistic writing has to say so. The first is that the elevation is entirely dosing-dependent. It is sustained only as long as the daily stimulus continues. Tesamorelin is not resetting a set point; it is repeatedly re-lifting IGF-1 against the constant downward pull of the body’s own regulation. When dosing stops, the driver disappears and the system returns toward baseline. Discontinuation studies in the HIV program make this concrete: the visceral-fat benefit — a downstream consequence of the GH/IGF-1 stimulation — re-accumulates after treatment is withdrawn, and by direct inference the IGF-1 elevation that produced it recedes as well.8 There is no evidence of a durable, treatment-free elevation.
The second qualification is that the elevation is physiologically bounded. Because tesamorelin works through the intact GHRH-R and leaves the somatotroph in charge, the rise in IGF-1 remains subject to the body’s negative-feedback machinery (the subject of the next section). This is arguably the compound’s single most important pharmacological feature and the sharpest contrast with injecting growth hormone directly. Exogenous GH bypasses pituitary regulation and can push GH and IGF-1 to supraphysiologic levels; tesamorelin, by acting one step upstream, produces an elevation that the system can still see and partially restrain.5 So “sustained” here means “maintained within a feedback-governed range for as long as dosing continues,” not “driven ever higher” and not “permanent.”
Holding those distinctions together, the most defensible one-sentence answer to the title is this: tesamorelin produces a sustained IGF-1 elevation in the sense that repeated daily GHRH-receptor stimulation, transduced through cAMP–PKA–CREB in the pituitary and GHR–JAK2–STAT5b in the liver and buffered by IGF-binding proteins, maintains a stable, feedback-limited increase in circulating IGF-1 for the duration of therapy — an increase that is neither supraphysiologic by design nor durable once dosing ends.
The Feedback Architecture That Keeps IGF-1 in Check
No account of “sustained” IGF-1 elevation is complete without the loops that prevent it from running away. The growth-hormone axis is one of the more elegantly regulated endocrine systems in the body, and tesamorelin’s upstream mechanism means it operates within that regulation rather than outside it.
Two feedback layers matter here. The first is short-loop feedback: growth hormone itself acts within the hypothalamus to increase the release of somatostatin (growth-hormone-inhibiting hormone) and to dampen GHRH release.5 Somatostatin then travels to the pituitary and directly opposes GHRH-driven secretion, effectively applying a brake proportional to how hard the accelerator has been pressed. The second layer is long-loop feedback from IGF-1: the very IGF-1 that GH stimulates in the liver circulates back to the hypothalamus and pituitary, where it suppresses further GH release — partly by stimulating somatostatin and partly by inhibiting GHRH and the somatotroph response.7 This long-loop suppression is a classic, well-documented negative-feedback arrangement: rising IGF-1 tells the system to ease off.7
The functional significance for tesamorelin is profound. Because the drug stimulates the pituitary rather than replacing its output, both feedback layers remain fully operational. As IGF-1 climbs in response to daily dosing, the long loop begins to restrain further GH secretion, and somatostatin tone rises to counter the GHRH-like push. The result is a self-limiting elevation: IGF-1 reaches a new, higher plateau at which the enhanced GHRH signaling is balanced by the enhanced feedback braking, and it tends to stay near that plateau rather than escalating without limit. This is the molecular reason tesamorelin’s IGF-1 rise is characterized as physiologic and feedback-preserved.5
The following table summarizes the difference between an upstream secretagogue like tesamorelin and direct hormone replacement, because that contrast is the crux of why the feedback architecture is preserved.
| Feature | Tesamorelin (GHRH analog, upstream) | Exogenous growth hormone (direct) |
|---|---|---|
| Site of action | Pituitary GHRH receptor4 | Peripheral GH receptors directly |
| GH secretion pattern | Pulsatile, somatotroph-controlled1 | Non-pulsatile, determined by injection |
| Short-loop feedback (somatostatin) | Intact and operative5 | Bypassed |
| Long-loop feedback (IGF-1 → hypothalamus/pituitary) | Intact; limits the elevation7 | Bypassed; can overshoot |
| Ceiling on IGF-1 | Feedback-bounded, tends to plateau5 | Can reach supraphysiologic levels |
| Reversibility on stopping | Returns toward baseline8 | Returns toward baseline |
This feedback-preserving design is the shared rationale across the GHRH-analog and secretagogue class, and it is why these compounds are often discussed as a group. For a broader view of how researchers frame this category and its handling, the site’s peptide glossary catalogs the relevant terminology, and the pillar discussion of sermorelin and the aging GH axis examines the same feedback logic in a different clinical context.
What the Clinical Trials Actually Measured
Mechanism is only as trustworthy as the data behind it, so it is worth being explicit about what the human trials of tesamorelin measured, in whom, and to what end — because those trials define the boundary of what can be claimed.
The evidence base is dominated by two pivotal, randomized, double-blind, placebo-controlled phase 3 trials in adults with HIV-associated lipodystrophy and excess abdominal fat. In the first, reported by Falutz and colleagues, 412 patients received 2 mg of tesamorelin or placebo by daily subcutaneous injection for 26 weeks. Visceral adipose tissue (VAT), measured by CT, fell by approximately 15% with tesamorelin while rising about 5% with placebo, and IGF-1 rose substantially in the treated group.2 A second, similarly designed pivotal trial with a safety extension replicated the core VAT and IGF-1 findings and provided longer-term follow-up.8 A pooled analysis of both trials plus extension data (roughly 800 participants) confirmed a VAT reduction on the order of 15% at 26 weeks that was maintained through 52 weeks of continued dosing, establishing the durability-during-treatment that underlies the “sustained” framing.6
Two later, mechanistically informative trials from Stanley and colleagues extended the picture to the liver. A 2014 randomized trial in HIV patients with abdominal fat accumulation confirmed reductions in visceral fat and documented modest reductions in liver fat.9 A subsequent randomized trial focused specifically on non-alcoholic fatty liver disease in HIV showed that tesamorelin reduced hepatic fat fraction and slowed fibrosis progression relative to placebo.11 Importantly, secondary analyses linked the metabolic improvements to the reduction in visceral adiposity rather than treating IGF-1 elevation as a benefit in itself.10 The table below lays out the core trial landscape.
| Trial / analysis | Population & design | Key IGF-1 / outcome finding |
|---|---|---|
| Falutz 2007 (pivotal 1) | 412 HIV patients, 26 wk, 2 mg/day vs placebo2 | VAT −15% vs +5%; significant IGF-1 rise |
| Falutz 2010 (pivotal 2 + extension) | HIV patients, RCT with safety extension8 | Replicated VAT reduction; IGF-1 elevation maintained |
| Falutz 2010 pooled (JCEM) | ~800 participants, two phase 3 trials pooled6 | ~15% VAT reduction; sustained through 52 wk of dosing |
| Stanley 2014 (JAMA) | 50 HIV patients, 6 mo, MGH9 | Reduced visceral fat; modest liver-fat reduction |
| Stanley 2019 (Lancet HIV) | 61 HIV patients with NAFLD, 12 mo11 | −37% relative hepatic fat; less fibrosis progression |
Two honest caveats frame this whole table. First, every one of these trials was conducted in people with HIV-associated fat accumulation — the approved population. None of them was a study of healthy adults, older adults seeking “anti-aging” effects, or athletes, and the IGF-1 and body-composition results cannot be assumed to transfer unchanged to those groups. Second, the endpoints that earned approval were fat-related (visceral adipose tissue, waist circumference), not IGF-1 per se. IGF-1 was measured as a pharmacodynamic marker confirming target engagement, and as a safety parameter to watch, not as a therapeutic goal. That ordering matters for the next section.
IGF-1 Elevation Versus Clinical Benefit: Keeping the Claims Honest
Here is where mechanistic enthusiasm most often outruns the evidence, so it deserves a section of its own. The pathway from tesamorelin to elevated IGF-1 is well established. It is tempting to leap from “IGF-1 is a powerful anabolic and metabolic hormone” to “therefore raising it with tesamorelin confers broad anabolic and metabolic benefits.” That leap is not supported by the tesamorelin data, and treating IGF-1 elevation as if it were the clinical prize is a category error.
What the trials actually demonstrated is narrow and specific: in adults with HIV-associated lipodystrophy, daily tesamorelin reduces excess visceral abdominal fat, an effect that is maintained during continued dosing and reverses on discontinuation.268 The liver-fat findings in the same population are a genuine and interesting extension of that metabolic effect.911 But the IGF-1 rise is best understood as the mechanism by which those fat effects occur — growth hormone and IGF-1 are lipolytic and influence visceral adipose metabolism — rather than as an independent benefit that has been shown to improve strength, cognition, longevity, or any other outcome people commonly associate with IGF-1. The published mechanistic work in HIV-associated NAFLD reinforces this framing: tesamorelin’s hepatic effects trace to GH-axis augmentation acting on defined transcriptomic pathways, and the benefit is metabolic and tissue-specific, not a generic consequence of “higher IGF-1.”12
This is the point at which off-label narratives most need discipline. Because tesamorelin reliably raises IGF-1, it is marketed in various non-medical channels for muscle gain, recovery, and anti-aging. The honest position is that these uses are unproven for tesamorelin specifically: there is no body of controlled trials showing that tesamorelin builds muscle, improves athletic performance, slows aging, or enhances cognition in any population, let alone in healthy adults. Raising a biomarker that is associated with those outcomes is not the same as demonstrating those outcomes, and the muscle-wasting literature is full of agents that moved a biomarker without moving function. Anyone reasoning from the IGF-1 mechanism to a promised benefit is filling an evidentiary gap with plausibility. The related question of whether a growth-hormone fragment can be assumed to deliver muscle benefits is examined honestly in the discussion of the clinical evidence around AOD-9604, another GH-derived compound whose real-world efficacy proved far more modest than its mechanism suggested.
None of this diminishes the legitimacy of the approved use. Within HIV-associated lipodystrophy, tesamorelin is a properly studied, FDA-approved therapy with real, replicated effects.1 The discipline lies in keeping the mechanistic story — sustained, feedback-bounded IGF-1 elevation — anchored to the outcomes that were actually measured, and in labeling everything beyond that as hypothesis.
The Kinetics of Sustained Elevation: Pulses, Plateaus, and Time Course
Returning to the pharmacodynamics with the pieces now in place, we can describe the time course of the IGF-1 elevation with some precision, because it is the interplay of fast and slow elements that produces the “sustained” appearance.
On the timescale of minutes, tesamorelin acts fast: subcutaneous absorption is rapid, GHRH-R activation triggers calcium-dependent GH exocytosis, and a GH pulse appears. On the timescale of hours, that GH pulse is transduced at the liver into IGF-1 synthesis via JAK2/STAT5b, and the newly made IGF-1 is captured into IGFBP-3/ALS ternary complexes that dramatically prolong its residence in the circulation.5 On the timescale of days, repeated once-daily dosing keeps re-triggering this sequence before the binding-protein reservoir has fully drained, so IGF-1 settles at an elevated steady state rather than oscillating back to baseline between injections. And on the timescale of weeks to months, the transcriptional replenishment of somatotroph GH stores, together with the intact feedback loops, keeps the system responsive and the plateau stable — which is exactly what the 52-week pooled data showed.6
Two properties of this multi-timescale system are worth emphasizing because they are frequently misunderstood. First, the plateau is a balance, not a ceiling imposed by drug saturation. IGF-1 rises until the added GHRH-like drive is matched by the added negative feedback, and then it holds. Increasing the dose does not linearly increase IGF-1 without limit, because the feedback loops tighten as IGF-1 climbs.7 Second, the plateau is dynamic and requires continuous input. Because IGF-1 and its binding proteins turn over, and because the feedback loops immediately sense any change, the elevated state is actively maintained by each day’s dose. Miss the doses and the balance shifts back toward baseline within a relatively short window — the same reversibility that manifests as visceral-fat re-accumulation after discontinuation.8
This is the most accurate mental model for “sustained”: a dynamic equilibrium held aloft by daily stimulation and constrained by intact feedback, not a permanent recalibration. It is sustained the way a fountain’s height is sustained by a running pump, not the way a filled reservoir stays full after the tap is closed. This distinction is not pedantry; it determines how the elevation should be interpreted in any research or clinical context. A value that reflects an actively maintained balance will move if the daily stimulus is interrupted, if the feedback set point shifts, or if the downstream binding-protein capacity changes — so a single IGF-1 measurement captures the state of the whole loop at one instant rather than a fixed pharmacological property of the drug. Understanding the elevation as a running equilibrium also clarifies why dose-titration against IGF-1, rather than a fixed dose chasing an ever-higher number, is the rational way to manage the approved therapy: the goal is to hold a bounded, feedback-consistent plateau, not to maximize a biomarker. Researchers documenting the practical handling parameters that keep such a daily-dosing regimen consistent will find the general principles of peptide preparation covered in the peptide reconstitution guide, offered strictly as educational reference rather than as instructions for human use.
Safety Implications of the IGF-1 Rise
Because sustained IGF-1 elevation is the pathway’s defining pharmacodynamic effect, the safety conversation is largely a conversation about the consequences of raising IGF-1 and growth hormone — and here, too, honesty requires acknowledging both the reassuring design features and the real cautions.
The reassuring feature, discussed above, is that tesamorelin’s upstream mechanism keeps the elevation within a feedback-bounded, physiologic range rather than pushing it to the supraphysiologic levels achievable with direct GH administration.5 In the pivotal trials, tesamorelin was generally well tolerated over the studied durations, and the metabolic effects were achieved without the dramatic overshoot in IGF-1 that unregulated GH dosing can produce.26 That said, several categories of concern are established or biologically expected, and the FDA labeling for Egrifta reflects them.1
- Glucose metabolism. Growth hormone is counter-regulatory to insulin, so any agent that raises GH and IGF-1 can affect glucose handling. Monitoring of glucose status is part of the labeled use, and the trials paid careful attention to this, generally finding the effect manageable within the studied populations but not absent.16
- Fluid retention and musculoskeletal symptoms. Arthralgia, myalgia, peripheral edema, and injection-site reactions were among the more common adverse events in the trials — effects consistent with GH-axis stimulation.2
- Theoretical neoplasia signal. Because IGF-1 has proliferative and anti-apoptotic activity, chronically elevating it raises a theoretical concern regarding neoplasia; the label directs that tesamorelin not be used in patients with active malignancy and that any elevation be weighed against that biology.1
- Contraindications and monitoring. Labeling contraindicates use in pregnancy and in disruption of the hypothalamic-pituitary axis (for example, from hypophysectomy or active tumor), and calls for IGF-1 monitoring during therapy precisely because the elevation is the intended effect that must be kept within reason.1
The overarching safety point mirrors the efficacy point: everything established about tesamorelin’s safety was established in adults with HIV-associated lipodystrophy over defined study periods. Short-term tolerability in that population does not license claims of long-term safety in healthy adults using the compound off-label for unapproved purposes, where the risk-benefit calculus is entirely different because there is no demonstrated benefit to weigh against the risks. A feedback-bounded IGF-1 rise is safer than an unbounded one, but “safer than the worst alternative” is not the same as “safe for any use.”
Where Tesamorelin Sits Among GH-Axis Strategies
Placing tesamorelin alongside neighboring approaches clarifies both what its pathway uniquely offers and where its limits lie. Broadly, there are three ways to raise IGF-1 through the GH axis, and they differ in exactly the feature that has occupied this article: how they interact with feedback.
The first strategy is direct growth hormone replacement, which bypasses the pituitary entirely and can drive IGF-1 to whatever level the dosing produces, feedback notwithstanding. The second is GHRH-based stimulation — tesamorelin, sermorelin, and related analogs — which acts at the GHRH receptor, preserves pulsatility and feedback, and produces a bounded elevation. The third is ghrelin-receptor (GHSR-1a) agonism — the growth-hormone secretagogues such as ipamorelin — which releases GH through a parallel receptor and is frequently discussed in combination with GHRH analogs because the two pathways can be complementary. Tesamorelin is distinctive within the second group for its DPP-4-resistant design and, importantly, for being the only member of the broader family with a full FDA approval backed by large phase 3 trials.12
The combination logic that pairs a GHRH analog with a GHSR-1a secretagogue — on the theory that stimulating two upstream inputs yields a larger GH pulse than either alone — is a common framing in the research-peptide literature and is explored in the discussion of the Grow-H blend and its claimed strength and repair effects. It is worth stressing that such combination approaches are largely exploratory and are not what tesamorelin was approved for; the approved use is tesamorelin monotherapy in a specific HIV population. For an organized overview of how these various GH-axis and metabolic compounds are catalogued for educational reference, the central dosages index groups them by mechanism and intended research context.
The comparative takeaway is that tesamorelin occupies a well-defined niche: an upstream, feedback-preserving, DPP-4-stabilized GHRH analog whose sustained IGF-1 elevation has been documented in rigorous trials, but only for a narrow approved indication. Its mechanism is arguably cleaner and more physiologic than direct GH; its evidence base outside HIV lipodystrophy is essentially absent. Both facts are true at once, and responsible communication holds them together.
Limitations, Open Questions, and the Boundary of the Evidence
Pulling the threads together, several limitations bound what can honestly be said about tesamorelin and sustained IGF-1 elevation.
Population specificity. The entire high-quality evidence base — the pathway’s pharmacodynamic confirmation, the durability during dosing, the safety profile — comes from adults with HIV-associated lipodystrophy.268 Whether the IGF-1 response, its magnitude, and its safety look the same in healthy or older adults is not established, and the feedback architecture that bounds the elevation could behave differently across differing baseline GH-axis states.
Biomarker versus benefit. IGF-1 elevation is a mechanism, not an outcome. The approved benefit is visceral-fat reduction; the extended benefits (liver fat, fibrosis) are within the same metabolic domain and the same HIV population.911 There is no controlled evidence that the IGF-1 rise itself delivers the muscle, performance, cognitive, or longevity benefits often attributed to it in off-label marketing. To date, no randomized controlled trial has tested tesamorelin for any of those outcomes in healthy or aging adults, so such claims remain investigational and unsupported by human data, and nothing in the approved evidence base should be read as validating them.
Reversibility. The elevation is dosing-dependent and reverses after discontinuation, which means any benefit derived from it is likewise transient and requires ongoing therapy to maintain — a genuine limitation for a chronic condition and a caution against imagining a one-time “reset.”8
Long-term consequences of chronic elevation. The theoretical neoplasia concern tied to IGF-1’s proliferative biology remains exactly that in the studied populations — theoretical and managed by contraindication — but the long-horizon consequences of years of maintained IGF-1 elevation are not fully characterized, and this uncertainty is greater, not lesser, in unstudied off-label use.1
The open scientific questions are correspondingly clear: how the pathway behaves across different baseline GH-axis states; whether the feedback-bounded elevation carries any of the broader benefits mechanistically hypothesized for it, tested in properly controlled trials rather than assumed; and what the true long-term safety of sustained elevation looks like beyond the durations studied. Until such work exists, the mechanistic account in this article should be read as a faithful description of how tesamorelin raises IGF-1, and emphatically not as an endorsement of raising IGF-1 for any purpose beyond the approved one. Readers tracking how this evidence base evolves across the peptide field can follow new literature as it appears.
Frequently Asked Questions
What is the core molecular pathway linking tesamorelin to IGF-1 elevation?
Tesamorelin binds the GHRH receptor on pituitary somatotrophs, a G-protein-coupled receptor that activates Gαs, adenylyl cyclase, and cAMP. Rising cAMP activates protein kinase A, which both triggers calcium-dependent release of stored growth hormone and phosphorylates CREB to upregulate Pit-1 and GH gene transcription.4 The released growth hormone then acts on hepatic GH receptors via the JAK2/STAT5b pathway to drive IGF-1 synthesis in the liver, the main source of circulating IGF-1.5 In short: GHRH-R → cAMP/PKA/CREB → GH release → hepatic JAK2/STAT5b → IGF-1.
In what sense is the IGF-1 elevation “sustained”?
In two legitimate senses: it holds a relatively even level between daily doses because IGF-binding proteins buffer the pulsatile GH signal, and it is maintained across many months of continued dosing without the pituitary becoming refractory, as shown in the pooled phase 3 data through 52 weeks.6 But it is sustained only while dosing continues and only within a feedback-bounded range — it is a dynamic equilibrium held up by daily stimulation, not a permanent new set point.
Does tesamorelin raise IGF-1 to dangerous, supraphysiologic levels?
Its design specifically avoids that. Because tesamorelin acts upstream at the pituitary, the body’s short-loop (somatostatin) and long-loop (IGF-1) negative feedback remain intact and restrain the elevation, so it tends to plateau within a physiologic range rather than overshooting the way direct growth hormone can.57 Nonetheless, IGF-1 monitoring is part of the approved labeling precisely because the elevation is the intended effect that must be kept in check.1
What is tesamorelin actually approved to treat?
Only one thing: the reduction of excess visceral abdominal fat in adults with HIV-associated lipodystrophy, marketed as Egrifta.1 Its efficacy for that indication rests on large randomized phase 3 trials showing roughly 15% reductions in visceral adipose tissue.26 All other uses — anti-aging, muscle building, athletic recovery, general metabolic optimization — are off-label and are not supported by controlled efficacy trials.
Does raising IGF-1 with tesamorelin build muscle or slow aging?
There is no controlled evidence that it does. IGF-1 is a biomarker associated with anabolic and metabolic processes, but tesamorelin was studied and approved for visceral-fat reduction, not for muscle, performance, cognition, or longevity outcomes.29 Reasoning from “IGF-1 is anabolic” to “tesamorelin builds muscle” substitutes mechanism for evidence, and that leap has not been tested for tesamorelin in any population.
What happens to IGF-1 when tesamorelin is stopped?
It recedes toward baseline. The elevation is entirely dosing-dependent, and the downstream visceral-fat benefit re-accumulates after discontinuation, indicating that the driving IGF-1 rise fades once the daily stimulus is withdrawn.8 There is no evidence of a durable, treatment-free elevation or a lasting “reset” of the axis.
How does tesamorelin differ from injecting growth hormone directly?
Tesamorelin stimulates the pituitary to make and release its own growth hormone in a pulsatile, feedback-controlled way, whereas exogenous GH bypasses the pituitary and its regulation entirely.15 The practical consequence is that tesamorelin’s IGF-1 elevation stays within a physiologic, self-limiting range, while direct GH can push IGF-1 to supraphysiologic levels because the feedback brakes are bypassed.
Why is tesamorelin resistant to breakdown when native GHRH is not?
Native GHRH is rapidly cleaved by the enzyme DPP-4 at its N-terminus. Tesamorelin adds a trans-3-hexenoyl group to the N-terminal tyrosine of the full GHRH(1–44) sequence, which sterically shields the vulnerable cleavage site while preserving receptor binding, extending the peptide’s functional half-life enough to support once-daily dosing.3
Is the liver the only place tesamorelin raises IGF-1?
The liver produces the great majority of circulating IGF-1 in response to growth hormone, so hepatic GHR/JAK2/STAT5b signaling is the dominant contributor to the measured systemic elevation.5 IGF-1 is also produced locally in many tissues under GH and other influences, but the sustained rise in blood IGF-1 that clinical studies track is primarily hepatic in origin.
References
- Theratechnologies Inc. EGRIFTA (tesamorelin for injection) U.S. Prescribing Information; FDA Center for Drug Evaluation and Research, NDA 022505. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022505Orig1s000SumR.pdf
- Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359-2370. PMID: 18057338. https://www.nejm.org/doi/full/10.1056/NEJMoa072375
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Tesamorelin. National Institute of Diabetes and Digestive and Kidney Diseases. Bookshelf ID: NBK548730. https://www.ncbi.nlm.nih.gov/books/NBK548730/
- Halmos G, Szabo Z, Dobos N, Juhasz E, Schally AV. Growth hormone-releasing hormone receptor (GHRH-R) and its signaling. Rev Endocr Metab Disord. 2025;26(3):343-352. PMCID: PMC12137518. https://pmc.ncbi.nlm.nih.gov/articles/PMC12137518/
- Steyn FJ, Tolle V, Chen C, Epelbaum J. Neuroendocrine regulation of growth hormone secretion. Compr Physiol. 2016;6(2):687-735. PMID: 27065166. https://onlinelibrary.wiley.com/doi/10.1002/j.2040-4603.2016.tb00692.x
- Falutz J, Mamputu JC, Potvin D, et al. Effects of tesamorelin (TH9507), a growth hormone-releasing factor analog, in HIV-infected patients with excess abdominal fat: a pooled analysis of two multicenter, double-blind placebo-controlled phase 3 trials with safety extension data. J Clin Endocrinol Metab. 2010;95(9):4291-4304. PMID: 20554713. https://academic.oup.com/jcem/article-abstract/95/9/4291/2835394
- Bermann M, Jaffe CA, Tsai W, DeMott-Friberg R, Barkan AL. Negative feedback regulation of pulsatile growth hormone secretion by insulin-like growth factor I. Involvement of hypothalamic somatostatin. J Clin Invest. 1994;94(1):138-145. PMID: 7913710. https://pubmed.ncbi.nlm.nih.gov/7913710/
- Falutz J, Potvin D, Mamputu JC, et al. Effects of tesamorelin, a growth hormone-releasing factor, in HIV-infected patients with abdominal fat accumulation: a randomized placebo-controlled trial with a safety extension. J Acquir Immune Defic Syndr. 2010;53(3):311-322. PMID: 20101189. https://pubmed.ncbi.nlm.nih.gov/20101189/
- Stanley TL, Feldpausch MN, Oh J, et al. Effect of tesamorelin on visceral fat and liver fat in HIV-infected patients with abdominal fat accumulation: a randomized clinical trial. JAMA. 2014;312(4):380-389. PMID: 25038357. https://pubmed.ncbi.nlm.nih.gov/25038357/
- Stanley TL, Falutz J, Mamputu JC, et al. Reduction in visceral adiposity is associated with an improved metabolic profile in HIV-infected patients receiving tesamorelin. Clin Infect Dis. 2012;54(11):1642-1651. PMID: 22495074. https://pubmed.ncbi.nlm.nih.gov/22495074/
- Stanley TL, Fourman LT, Feldpausch MN, et al. Effects of tesamorelin on non-alcoholic fatty liver disease in HIV: a randomised, double-blind, multicentre trial. Lancet HIV. 2019;6(12):e821-e830. PMID: 31611038. https://www.thelancet.com/journals/lanhiv/article/PIIS2352-3018(19)30331-5/abstract
- Fourman LT, Billingsley JM, Agyapong G, et al. Effects of tesamorelin on hepatic transcriptomic signatures in HIV-associated NAFLD. JCI Insight. 2020;5(16):e140134. PMCID: PMC7455119. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7455119/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. Tesamorelin (Egrifta) is approved by the U.S. Food and Drug Administration only for the reduction of excess visceral abdominal fat in adults with HIV-associated lipodystrophy; it is not approved for anti-aging, muscle building, athletic performance, general metabolic enhancement, or any other use, and no controlled trials support those off-label applications. The IGF-1 elevation described here is a pharmacodynamic mechanism, not a demonstrated clinical benefit outside the approved indication. Nothing here is medical advice or a recommendation for human use. Tesamorelin affects the growth hormone axis and carries glucose, fluid-retention, and theoretical neoplasia considerations; any legitimate use should occur under qualified medical supervision and in accordance with its approved labeling and applicable regulations.