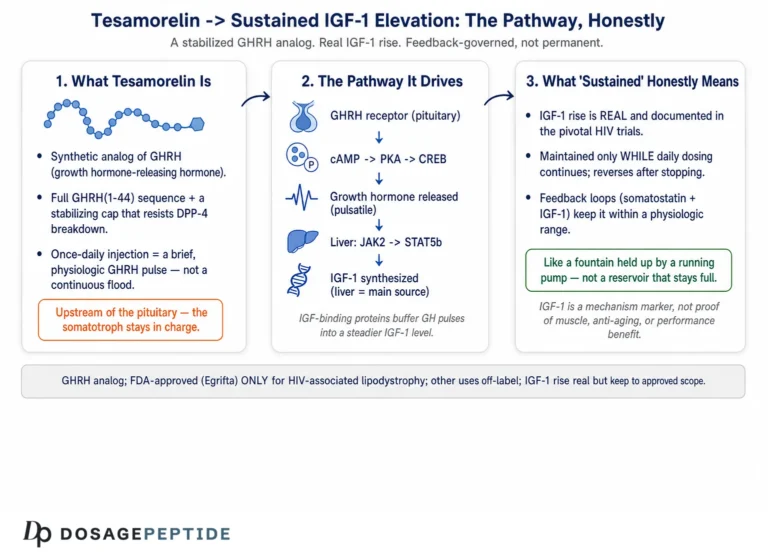
The question in the title is, refreshingly, a mechanistic one rather than a marketing one. It does not ask whether tesamorelin is a fountain of youth or a body-recomposition shortcut; it asks how a chain of molecular signaling events converts a once-daily subcutaneous injection into a measurable rise in circulating insulin-like growth factor 1 (IGF-1). That is a fair and answerable question, because the pathway is genuinely well characterized — tesamorelin is one of the few peptides in this space whose mechanism was worked out in registrational human trials rather than inferred from rodent data and vendor copy. It is approved by the U.S. Food and Drug Administration under the brand name Egrifta for one narrow purpose: to reduce excess visceral abdominal fat in people with HIV-associated lipodystrophy.1 The IGF-1 elevation the title refers to is real, reproducible, and central to how the drug works.2
But a mechanistic question still carries an implicit framing worth flagging before we start. Describing tesamorelin as something that “drives IGF-1 elevation” can slide, in the reader’s mind, into the assumption that raising IGF-1 is itself the therapeutic goal — that more IGF-1 is simply better. That is not how the approved science reads. IGF-1 elevation is the biochemical signature that the growth-hormone axis has been engaged; it is a biomarker of target engagement and, in the approved population, a mediator of visceral-fat loss. It is emphatically not a free-standing benefit to be maximized. In fact, the same IGF-1 rise that signals the drug is working is also the parameter clinicians are told to monitor and, if it climbs too high, the trigger to stop treatment.1 So the honest way to read the title is: let us trace the signaling network faithfully, and in doing so understand why the IGF-1 output is both the point of the drug and one of its most important guardrails.
This article is written for researchers and scientifically literate readers who want an accurate map of the cascade — from the amino-acid design of the molecule, through its receptor on the pituitary somatotroph, out into the pulsatile release of growth hormone (GH), down to the hepatic GH receptor and the JAK2/STAT5b machinery that transcribes the IGF1 gene, and back again through the feedback loops that keep the whole system in check. Along the way we will stay disciplined about scope: where a claim rests on the pivotal HIV-lipodystrophy trials, we will say so; where a use is off-label or investigational, we will label it plainly; and we will not let the elegance of the mechanism inflate the modest, specific set of things tesamorelin has actually been shown to do.
What Tesamorelin Is and Why Its Molecular Design Matters
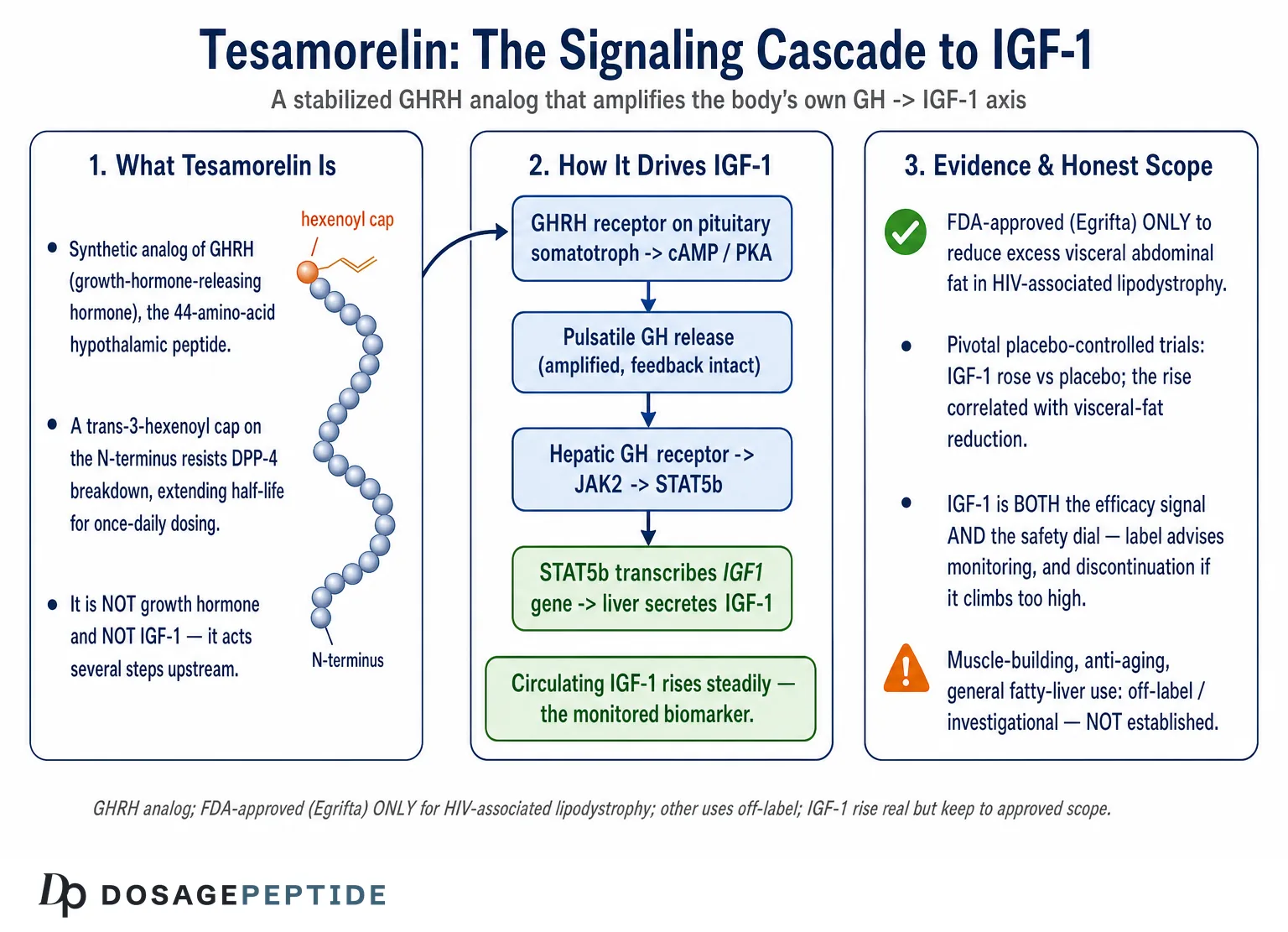
Tesamorelin, known in development as TH9507, is a synthetic analog of human growth-hormone-releasing hormone (GHRH), the 44-amino-acid hypothalamic peptide that normally instructs the pituitary to make and release growth hormone. The molecule reproduces the full biologically active GHRH(1–44) sequence and adds a single, deliberate modification: a trans-3-hexenoyl group is attached to the N-terminal tyrosine.3 That small acyl cap is the entire engineering story, and it is worth understanding because it explains why tesamorelin behaves as a practical drug where native GHRH does not.
Native GHRH is exquisitely short-lived in plasma. Its N-terminus is the business end — the residues that dock into and activate the receptor — and it is also the region most rapidly chewed apart by the enzyme dipeptidyl peptidase-4 (DPP-4), which clips off the first two residues and abolishes activity within minutes. The trans-3-hexenoyl modification sterically shields that vulnerable N-terminus, slowing enzymatic degradation and extending the peptide’s functional half-life enough to make once-daily subcutaneous dosing pharmacologically meaningful.3 Crucially, the modification does not change what the molecule does at the receptor; it changes how long it survives to do it. Tesamorelin remains a full agonist at the GHRH receptor. This is the first and most important design fact for understanding the IGF-1 question: tesamorelin is not a growth hormone and not an IGF-1 mimetic. It is a stabilized upstream trigger that acts several signaling nodes before IGF-1 ever appears.
That distinction separates tesamorelin from two other classes of compound it is frequently confused with. It is not exogenous recombinant growth hormone, which floods the circulation with GH directly and bypasses the pituitary altogether. And it is not a ghrelin-mimetic growth-hormone secretagogue such as ipamorelin, which acts on a completely different receptor (GHS-R1a) to release GH by a parallel route; researchers comparing the two mechanisms can see how the GHS-R1a arm is characterized in the discussion of ipamorelin’s receptor targeting. Tesamorelin belongs instead with the GHRH-analog family, whose closest studied relative is sermorelin, GHRH(1–29); the way that shorter GHRH fragment stimulates endogenous GH is described in the site’s overview of sermorelin’s role in stimulating natural growth hormone. What sets tesamorelin apart within even that family is that it carries the full 1–44 sequence with the stabilizing cap, and that it alone has been carried through pivotal, placebo-controlled human efficacy trials to regulatory approval.1
Holding these categories distinct is the single most useful habit for reasoning about the IGF-1 output. Because tesamorelin sits so far upstream, every downstream effect it produces — including the IGF-1 rise — is filtered through the pituitary’s own regulatory logic. That filtering is not a bug; it is the feature that makes the compound behave more like an amplifier of a physiological rhythm than like a hormone override. The table below situates tesamorelin among the classes it is often conflated with, and the differences are not cosmetic — they determine where in the signaling network each agent acts, and therefore how the resulting IGF-1 elevation is shaped and controlled.
| Agent / class | Site of action | Relationship to endogenous feedback | Human evidence status |
|---|---|---|---|
| Tesamorelin (GHRH(1–44) analog) | Pituitary GHRH receptor | Amplifies pulsatile GH; feedback intact3 | FDA-approved for HIV lipodystrophy1 |
| Sermorelin (GHRH(1–29)) | Pituitary GHRH receptor | Amplifies pulsatile GH; shorter, less stable ligand | Historically approved as a diagnostic; other uses off-label |
| Ghrelin-mimetic secretagogues (e.g., ipamorelin) | GHS-R1a receptor (parallel node) | Amplifies GH via a distinct pathway; feedback intact | Investigational; no approval for this use |
| Recombinant human GH | Bypasses pituitary entirely | Flat, non-pulsatile exposure; overrides feedback | Approved for defined GH-deficiency indications |
The consequential column is the third one. Only the agents that act at or above the pituitary leave the body’s feedback loops operating; recombinant GH does not, which is why its IGF-1 elevation is governed by injection pharmacokinetics rather than by the somatotroph’s own restraint. Tesamorelin’s approval status in the fourth column is equally worth internalizing: of these classes, it is the GHRH analog that has actually been carried to a regulatory endpoint, and even then for a single, specific population.
The First Node: GHRH Receptor Engagement on the Somatotroph
The signaling network begins where tesamorelin first makes molecular contact: the growth-hormone-releasing-hormone receptor (GHRH-R) on the surface of somatotroph cells in the anterior pituitary. The GHRH-R is a class B1 (secretin-family) G-protein-coupled receptor, a seven-transmembrane protein with a large extracellular N-terminal domain that captures the C-terminal helix of the peptide ligand while the ligand’s own N-terminus inserts into the transmembrane core to switch the receptor on.4 This two-domain “affinity-trap-and-activate” binding mode is characteristic of the class B GPCRs and explains why the N-terminal integrity of tesamorelin — the region the hexenoyl group protects — is so essential to agonism.
Once tesamorelin engages the receptor, the somatotroph runs a canonical class B signaling program. The activated GHRH-R couples predominantly to the stimulatory G protein Gαs, which activates adenylyl cyclase and raises intracellular cyclic AMP (cAMP).4 Rising cAMP activates protein kinase A (PKA), and PKA does two things that matter for GH output. First, it phosphorylates ion channels and promotes calcium influx, depolarizing the cell and triggering the immediate, regulated exocytosis of GH-containing secretory granules — this is the acute release of stored growth hormone that follows a dose within minutes to hours. Second, on a slower timescale, PKA phosphorylates the transcription factor CREB (cAMP-response-element-binding protein), and phosphorylated CREB drives transcription of the GH1 gene and helps sustain expression of the pituitary-specific transcription factor Pit-1 (POU1F1), which is required for somatotroph identity, proliferation, and continued GH synthesis.4
The result is a two-tier response that is easy to miss if one thinks of GHRH agonism as a simple “release” signal. Tesamorelin both empties the ready-releasable pool of GH (the acute secretory burst) and replenishes it by promoting new GH synthesis and somatotroph trophic support (the sustained transcriptional arm). Over weeks of daily dosing, this combination raises the amplitude of the body’s own GH pulses without the pituitary ever being taken offline. That is the mechanistic seed of everything downstream: a stronger but still endogenous GH signal, generated by the patient’s own somatotrophs rather than infused from a vial.
It is worth emphasizing what the GHRH-R does not do here. It does not itself raise IGF-1. No hepatocyte has responded yet; no IGF1 gene has been transcribed. At this first node, the only currency is growth hormone. The IGF-1 elevation the title asks about is still two organ systems and several signaling steps away, and it depends entirely on what happens when that pituitary-derived GH reaches its own target tissues.
There is a further consequence of acting at the GHRH receptor that bears on the IGF-1 output: the somatotroph population itself is finite and regulated. Tesamorelin can only mobilize and synthesize GH within the capacity of the patient’s own pituitary. In a person with an intact, healthy somatotroph reserve, that capacity is considerable, and the IGF-1 response is robust. In someone with pituitary insufficiency, prior hypophysectomy, or hypothalamic-pituitary disruption from other causes, the same dose of tesamorelin would produce a blunted or absent response, because there is no downstream GH to amplify — and this is precisely why such patients fall outside the drug’s approved use.1 The mechanism, in other words, has a built-in ceiling set by the biology of the responding cell, which is not true of an exogenous hormone infusion that simply raises the plasma level regardless of glandular reserve. This ceiling is one of the reasons the IGF-1 elevation produced by a GHRH analog tends to be self-limiting rather than open-ended.
Why Pulsatility Is the Whole Point: Secretagogue Versus Exogenous Hormone
A network diagram of tesamorelin’s action is incomplete without the concept of pulsatility, because it is the feature that distinguishes engaging the axis from overriding it. Physiological GH secretion is not a steady drip; it is released in discrete bursts, predominantly during slow-wave sleep, separated by troughs during which GH falls to nearly undetectable levels.5 This pulsatile pattern is not incidental. Target tissues read the pattern of GH exposure, not merely the average concentration: the peaks drive certain gene programs while the intervening troughs allow receptors to reset and prevent the desensitization that continuous exposure would cause.5
Because tesamorelin works by prompting the pituitary to release its own GH, it amplifies these endogenous pulses rather than replacing them with a flat, supraphysiological plateau.3 The somatotroph still answers to hypothalamic somatostatin, which periodically brakes secretion, and to the negative feedback exerted by GH and IGF-1 themselves. The consequence is a GH profile that is higher in amplitude but still recognizably rhythmic and still subject to shutdown. This is the mechanistic argument — and it is a mechanistic argument, not a proven clinical-superiority claim — for why a GHRH analog might produce a more “physiological” downstream signature than an equivalent exposure to injected recombinant GH, which delivers hormone on the pharmacokinetics of the injection rather than the biology of the pituitary.
The practical relevance for the IGF-1 question is subtle but important. IGF-1 is produced largely in the liver in response to GH, and because IGF-1 has a long circulating half-life (it travels bound to IGF-binding proteins and the acid-labile subunit), the pulsatile GH signal is effectively integrated into a smoother, more stable IGF-1 concentration. In other words, the spiky upstream GH signal is low-pass-filtered by hepatic IGF-1 biology into the steady rise clinicians measure in a morning blood draw. This is why IGF-1, rather than GH itself, is the biomarker used to track tesamorelin’s effect and safety: a single GH level is nearly meaningless given the pulses, whereas IGF-1 reflects the time-averaged strength of the whole axis.1
The same pulsatility logic is what motivates interest in other endogenous-GH strategies across the peptide field. Ghrelin-receptor secretagogues, GHRH fragments such as sermorelin, and combination approaches are all, in principle, attempts to raise GH output while keeping the pituitary in the loop; the broader family and how these compounds are catalogued for research reference appears in the site’s central dosages index. Tesamorelin is distinguished within that group less by a unique mechanism than by the depth of its human evidence in one specific indication.
From Pituitary to Liver: the GH Receptor, JAK2, and STAT5b
Now we reach the node the title is really about — the molecular machinery that converts elevated growth hormone into elevated IGF-1. This step happens mostly in the liver, though it also occurs locally in many peripheral tissues, and it runs through one of the best-characterized cytokine-receptor signaling systems in endocrinology.
Growth hormone circulates and binds the growth-hormone receptor (GHR), a single-pass transmembrane receptor that exists at the cell surface as a preformed dimer. GH binding does not simply glue two receptors together; rather, it induces a conformational rotation of the two receptor subunits relative to one another. This rotation repositions the intracellular domains and, with them, the Janus kinase 2 (JAK2) molecules that sit constitutively bound to each receptor tail via its Box1 motif.6 The reorientation relieves an inhibitory arrangement and brings the two JAK2 kinases into a productive geometry, allowing them to trans-phosphorylate and activate one another. This activation-by-realignment model, worked out over the last two decades, replaced the older “GH just dimerizes the receptor” picture and is the current consensus for how the GHR fires.6
Activated JAK2 then phosphorylates tyrosine residues on the GHR’s own cytoplasmic tail, creating docking sites. The most consequential recruits are the STAT proteins — signal transducers and activators of transcription — and for the IGF1 gene the critical one is STAT5b. JAK2 phosphorylates receptor-docked STAT5b, which then dissociates, dimerizes with a partner STAT5 molecule, and translocates into the nucleus.6 There it binds GH-response elements in and around target genes and directly activates transcription. Genetic evidence anchors this pathway firmly to IGF-1: STAT5b is required for GH-dependent IGF1 transcription, and humans with loss-of-function STAT5B mutations present with severe GH-insensitive growth failure and very low IGF-1 despite normal or high GH — the endocrine mirror image of what tesamorelin is trying to do.7
STAT5b is not the only output. Activated JAK2 also seeds the RAS/MAPK and PI3K/Akt cascades from the same receptor complex, which contribute to GH’s effects on cell proliferation, survival, and intermediary metabolism — including aspects of lipid and glucose handling that matter for tesamorelin’s fat-reducing action and for its metabolic side effects.6 But for the specific question of IGF-1 elevation, the JAK2→STAT5b→IGF1-transcription axis is the throughline. When tesamorelin raises pulsatile GH, more GH reaches hepatic GHRs, more JAK2 fires, more STAT5b reaches the nucleus, and the IGF1 gene is transcribed more vigorously. The liver secretes the resulting IGF-1 into the circulation, where it accumulates on its binding proteins into the stable elevation measured in trials.7
It is worth pausing on the timescales embedded in this step, because they explain the shape of the IGF-1 curve clinicians observe. The proximal events — receptor rotation, JAK2 autophosphorylation, STAT5b phosphorylation and nuclear entry — occur within minutes of a GH pulse arriving at the hepatocyte. Transcription of the IGF1 gene, translation, post-translational assembly of the ternary complex with IGFBP-3 and the acid-labile subunit, and secretion into the circulation add hours. Because the circulating ternary complex has a half-life measured in hours rather than minutes, IGF-1 accumulates and decays far more slowly than the GH pulses that drive it, and layered daily dosing pushes the system to a new plateau over one to two weeks. This hierarchy of timescales — fast signaling, slower transcription, slow clearance — is why a single injection does not produce a matching IGF-1 spike, and why the biomarker that reports on tesamorelin’s activity is a smoothed integral of many pulses rather than a snapshot of any one.7
Two features of this transcriptional step deserve emphasis. First, it is genuinely a gene-expression event, not a simple stoichiometric conversion of GH into IGF-1; the magnitude of the IGF-1 response therefore depends on hepatic health, nutritional and insulin status (portal insulin permissively supports hepatic GHR expression), thyroid status, and the integrity of the STAT5b machinery. This is part of why IGF-1 responses vary between individuals even at an identical tesamorelin dose. Second, because the same STAT5b pathway governs IGF-1 production in normal physiology, tesamorelin is not creating an artificial signal — it is turning up the gain on the body’s native GH→IGF-1 circuit. That is reassuring mechanistically and also precisely why the output must be monitored: the drug amplifies a real growth-signaling pathway with real biological consequences.
The IGF-1 Output: Magnitude, Kinetics, and What the Trials Actually Measured
Having traced the cascade, we can now put numbers on its endpoint, drawing strictly on the registrational human data rather than on extrapolation. In the pivotal placebo-controlled trials that supported approval, once-daily subcutaneous tesamorelin 2 mg produced a clear, reproducible rise in serum IGF-1 relative to placebo.2 Pooled analysis of the two phase 3 trials in HIV-infected adults with excess abdominal fat quantified this: IGF-1 rose substantially in the tesamorelin arm while remaining essentially flat on placebo, and — the mechanistically satisfying part — the degree of IGF-1 increase correlated with the degree of visceral-fat reduction, exactly as one would predict if the GH/IGF-1 axis were the effector pathway.8
The kinetics follow the biology described above. GH rises acutely after each dose, but IGF-1, integrating the pulses, climbs over days to weeks toward a new steady state and is then maintained with continued daily dosing; the pooled phase 3 program with its safety extension showed the IGF-1 elevation and the visceral-fat effect persisting through roughly a year of continuous treatment rather than waning through tachyphylaxis.8 Conversely, because tesamorelin does not create a depot and the axis is under active feedback, stopping the drug allows IGF-1 to fall back toward baseline and the visceral fat to re-accumulate — a reminder that the compound suppresses nothing permanently and rewrites no set-point; it maintains an amplified signal only for as long as it is given.8
| Signaling node | Molecular event | Measurable / observable output |
|---|---|---|
| Tesamorelin (TH9507) | Stabilized GHRH(1–44) analog; trans-3-hexenoyl cap resists DPP-4 cleavage3 | Longer functional half-life; once-daily dosing feasible |
| GHRH receptor (somatotroph) | Class B GPCR → Gαs → adenylyl cyclase → cAMP → PKA → CREB/Pit-14 | Acute GH exocytosis + sustained GH synthesis |
| Pituitary GH release | Amplified pulsatile secretion under intact somatostatin/feedback control5 | Higher-amplitude endogenous GH pulses |
| Hepatic GH receptor | GH-induced receptor rotation activates paired JAK26 | JAK2 autophosphorylation; docking-site creation |
| JAK2 → STAT5b | STAT5b phosphorylation, dimerization, nuclear translocation, IGF1 transcription7 | Increased hepatic IGF-1 synthesis and secretion |
| Circulating IGF-1 | IGF-1 stabilized on IGFBP-3 / acid-labile subunit2 | Steady, dose-related IGF-1 elevation (the monitored biomarker) |
The table makes the throughline explicit, but it also makes an honest point by omission: every row is a step at which the signal can be modulated, attenuated, or fed back upon. The IGF-1 elevation at the bottom is not a lever the drug pulls directly; it is the emergent output of a regulated multi-organ circuit. That is why the same 2 mg dose yields different IGF-1 levels in different people, and why IGF-1 monitoring — not dose alone — governs safe use.
Feedback Architecture: How the Network Regulates Its Own IGF-1 Output
A signaling network that only pushed in one direction would be dangerous. The GH/IGF-1 axis is instead ringed with negative-feedback loops, and understanding them is essential to understanding both why tesamorelin’s IGF-1 elevation is self-limiting and why it nonetheless requires vigilance.
The dominant brake is IGF-1 itself. Circulating IGF-1, along with GH, feeds back on the hypothalamus to stimulate somatostatin (the inhibitory counterpart of GHRH) and on the pituitary to blunt somatotroph responsiveness.5 As tesamorelin drives IGF-1 upward, this rising IGF-1 tends to restrain further GH secretion, so the system approaches a new, higher steady state rather than spiraling. Within cells, a second layer of control operates through the suppressor-of-cytokine-signaling (SOCS) proteins: GH signaling through JAK2/STAT5b induces SOCS expression, and SOCS proteins then dampen JAK2 activity and target the receptor complex for turnover, providing intracellular negative feedback that limits how strongly any given GH pulse can be transduced.6 These loops together explain the plateau seen in the trials: IGF-1 rises to a new level and holds, rather than climbing without bound, because the network resists its own amplification.
This feedback architecture also clarifies the contrast with parallel secretagogue strategies. Ghrelin-receptor agonists act at GHS-R1a, a distinct node that synergizes with GHRH signaling at the somatotroph and is subject to its own regulation; the receptor-level specifics are laid out in the discussion of ipamorelin and the GHS-R1a receptor. Combining a GHRH analog with a ghrelin-mimetic can, in principle, produce a larger GH pulse than either alone, which is one rationale behind investigational GH-axis blends; the strength-and-repair claims sometimes attached to such combinations are examined in the site’s coverage of the Grow-H blend. The key honest caveat is that stacking secretagogues also stacks the downstream IGF-1 drive, and the same feedback loops that make single-agent tesamorelin self-limiting can be overwhelmed — which is precisely the scenario in which IGF-1 monitoring becomes non-negotiable and which takes such combinations well outside any approved use.
The clean way to summarize the feedback story is this: tesamorelin does not disable the axis’s safety systems; it works within them. The IGF-1 elevation it produces is the output of a servo-controlled loop that is trying, at every node, to return toward its set-point. That is what makes the drug’s effect controllable, and it is also why the effect evaporates when dosing stops.
Translating IGF-1 Elevation Into the Approved Effect: Visceral Fat
None of this cascade would matter clinically if the IGF-1 elevation did not do something useful, and here we must be scrupulous about scope. The one effect for which tesamorelin has pivotal, placebo-controlled human evidence and regulatory approval is the reduction of excess visceral adipose tissue (VAT) in people with HIV-associated lipodystrophy.1 In the pivotal NEJM trial, once-daily tesamorelin reduced VAT significantly versus placebo over 26 weeks, with concomitant improvement in triglycerides and the total-to-HDL cholesterol ratio and, importantly, no significant worsening of the primary glucose measures over that window.2 The pooled phase 3 analysis put the VAT reduction on the order of roughly 15% versus placebo and confirmed the IGF-1–to–VAT-response relationship that ties the fat loss to the GH/IGF-1 mechanism.8
Mechanistically, the fat-loss effect is best understood as a GH-driven action to which IGF-1 is both a contributor and a marker. Growth hormone is directly lipolytic in adipose tissue — it promotes hydrolysis of stored triglyceride and shifts substrate use toward fat oxidation — and visceral fat appears particularly responsive to this signal. The IGF-1 arm supports the anabolic and metabolic context in which that lipolysis occurs. The net result in the approved population is a preferential loss of the metabolically active visceral depot rather than a general weight-loss effect; tesamorelin is not an obesity drug and was not studied or approved as one.1 This specificity — visceral fat in a defined patient group, driven by a defined hormonal axis — is exactly the kind of narrow, evidence-anchored claim that distinguishes tesamorelin’s approved profile from the sweeping body-composition promises that circulate around GH-axis peptides generally.
The signaling logic also clarifies why visceral fat, specifically, is the tissue that responds. Visceral adipocytes are densely innervated, highly lipolytically active, and rich in GH and beta-adrenergic signaling relative to subcutaneous depots, so an amplified GH pulse preferentially mobilizes triglyceride from exactly the compartment that is pathologically expanded in lipodystrophy. The IGF-1 arm, meanwhile, helps preserve lean tissue and supports the anabolic milieu, which is part of why the net body-composition shift in the trials favored visceral-fat loss without the indiscriminate lean-mass depletion that aggressive caloric restriction would produce. This is a coherent, mechanism-consistent story — and it is important precisely because it is bounded. The trials did not show, and the mechanism does not imply, that tesamorelin melts subcutaneous fat, produces cosmetic slimming, or drives weight loss in people without the lipodystrophic pattern of visceral accumulation. The specificity is the science, and it is the first thing lost when the compound is discussed as a generic fat-burner.
It bears repeating that HIV-associated lipodystrophy is a specific pathological state — a treatment-and-disease-related redistribution of body fat with a characteristic visceral accumulation and often adverse metabolic consequences — and that the approval, the dosing, and the risk-benefit judgment all pertain to that population. Extending the visceral-fat finding to healthy adults seeking aesthetic fat loss is not supported by the registrational evidence and moves the compound into off-label territory, where the favorable trial risk-benefit balance can no longer be assumed.
Off-Label and Investigational Directions: Reading the Premise Honestly
Because tesamorelin reliably engages the GH/IGF-1 axis, it has attracted research interest well beyond its approved indication. Honesty requires drawing a bright line here: these are investigational or off-label uses, several with genuine early data, none of which has produced a new FDA approval, and none of which should be read as an established therapy.
The most substantive investigational direction concerns the liver. Because visceral adiposity and ectopic liver fat travel together, and because GH signaling influences hepatic lipid handling, tesamorelin was tested for effects on hepatic fat in HIV. A randomized clinical trial reported that tesamorelin reduced liver fat and limited progression of fibrosis markers in HIV-infected patients with abdominal fat accumulation,9 and a subsequent randomized, double-blind, multicenter trial specifically in HIV patients with non-alcoholic fatty liver disease found that tesamorelin reduced hepatic fat fraction relative to placebo.10 These are real, peer-reviewed randomized findings — but note the tight framing: they are in people with HIV, they concern a biomarker (liver-fat fraction and histologic indices) rather than hard clinical liver outcomes, and they have not translated into an approved hepatic indication. They refine our understanding of what the GH/IGF-1 amplification does to ectopic fat; they do not license tesamorelin as a treatment for fatty liver disease in the general population.
A second cluster of claims travels under the banners of muscle-building, athletic recovery, and general anti-aging, and here the honest verdict is blunter still: there are no adequate, well-controlled human trials demonstrating that tesamorelin builds skeletal muscle, improves strength or physical performance, or extends healthspan, and it is not approved for any of these purposes. The registrational program never measured strength, power, endurance, or functional outcomes as endpoints; it measured visceral fat, lipids, glucose, and IGF-1 in a single disease population.8 The inference that a compound which raises IGF-1 must therefore be anabolic for muscle is a mechanism-to-outcome leap of exactly the kind this article is trying to discourage — biologically plausible in the abstract, but unvalidated for the specific claim. IGF-1 is indeed involved in muscle protein synthesis in physiology, yet plausibility is not evidence, and the marketing that surrounds GH-axis peptides routinely collapses that distinction. Anyone encountering tesamorelin framed as a performance or longevity agent is reading past the data, not from it.1
It is also worth stating plainly what has not been done: there are no head-to-head trials establishing that tesamorelin’s more “physiological” pulsatile IGF-1 profile produces better clinical outcomes than recombinant GH, no long-term oncological safety studies powered to resolve the theoretical malignancy question, and no trials in healthy, non-lipodystrophic adults that would justify the aesthetic or wellness uses for which the compound is informally sought. The absence of these studies is not a minor footnote; it is the boundary of what can honestly be claimed. Where the mechanism is elegant and the biomarker moves reliably, it is tempting to assume the clinical benefits follow — but the discipline the title’s mechanistic framing demands is precisely to hold that assumption in check until controlled human evidence supplies it.
Other directions are more speculative still. The role of the GH/IGF-1 axis in cognition and the age-related decline of that axis has prompted interest in GHRH analogs for brain aging, an area explored for the related compound in the site’s discussion of sermorelin and cognitive function in age-related neurodegeneration; for tesamorelin specifically, any cognitive or anti-aging application remains investigational and unproven, and the enthusiasm often outruns the data. The core discipline for reading all of these premises is the same one the title itself invites: trace the mechanism honestly, and then ask whether a mechanism-level plausibility has actually been converted into controlled human evidence for the specific claim being made. For tesamorelin, that conversion has happened for HIV-associated visceral fat and, in a narrower biomarker sense, for liver fat in HIV — and essentially nowhere else.
Safety, IGF-1 Monitoring, and the Limits of the Signal
The same feature that makes tesamorelin work — a genuine, sustained elevation of the GH/IGF-1 axis — is the source of its principal risks, which is why the mechanism and the safety profile cannot be discussed separately. Because IGF-1 is a growth and survival factor, and because chronically elevated IGF-1 has been epidemiologically associated with certain malignancies, the prescribing information carries specific cautions: tesamorelin is contraindicated in the presence of active malignancy, and it is not to be used in patients with disruption of the hypothalamic-pituitary axis from other causes.1 Its known effect on IGF-1 has an unknown effect on the development or progression of cancers, and this uncertainty — not a demonstrated harm, but an unresolved theoretical risk — shapes the whole risk-benefit calculus.1
The IGF-1 elevation is therefore not merely the efficacy signal; it is the safety dial. The label directs measurement of IGF-1 at baseline and periodically during treatment, and it advises considering discontinuation if IGF-1 rises persistently and substantially — particularly beyond about 3 standard deviations above the age- and sex-adjusted normal range — especially when the clinical benefit is not robust.1 This is a striking design principle: the biomarker that tells you the drug is engaging its target is the same one that, past a threshold, tells you to stop. It reframes the title’s premise usefully — the goal is not to “drive IGF-1 elevation” as high as possible, but to raise it into a therapeutic window and hold it there under surveillance.
Beyond the IGF-1-specific concerns, the metabolic consequences of GH amplification round out the safety picture. Growth hormone is counter-regulatory to insulin, so GH-axis activation can worsen insulin sensitivity and glucose tolerance; monitoring of glucose and glycated hemoglobin is advised, with particular attention in patients who have or are at risk for diabetes.11 Fluid retention, arthralgia, peripheral edema, carpal-tunnel-type symptoms, and injection-site reactions reflect the familiar spectrum of GH-mediated effects and are the commonly reported adverse events.11 Hypersensitivity reactions have also been reported. A meta-analysis of the randomized tesamorelin trials in HIV-associated lipodystrophy broadly confirmed the efficacy on visceral fat alongside this manageable-but-real adverse-event profile, and reinforced that the benefits were demonstrated in that specific population under monitoring.12
The honest safety synthesis is that tesamorelin’s risks are the predictable consequences of doing exactly what its mechanism says it does. There is no free lunch in which the axis is amplified for fat loss while the growth-and-metabolism consequences of that amplification are somehow avoided. In the approved, monitored HIV-lipodystrophy setting, the trials judged this balance acceptable. Outside that setting — unmonitored, in different populations, or stacked with other secretagogues — the same IGF-1 elevation that defines efficacy becomes an unsupervised and potentially hazardous signal.
Research Handling, IGF-1 Assays, and Measurement Discipline
For readers approaching tesamorelin in a laboratory or research context rather than a clinical one, a brief and strictly educational note on handling and measurement is warranted, with the standing caveat that this is standard research-peptide practice and not a usage recommendation, and that tesamorelin is a prescription drug for a single approved indication.
Tesamorelin is supplied as a lyophilized powder that is reconstituted with an appropriate sterile diluent for research use; as with other labile peptides, the diluent is directed gently against the vial wall rather than sprayed onto the cake, the vial is swirled rather than shaken to avoid shearing and foaming, and reconstituted material is kept cold, protected from light, and spared repeated freeze-thaw cycles that degrade peptide integrity. General principles of dissolving and stabilizing lyophilized peptides are laid out in the site’s peptide reconstitution guide, and unfamiliar terms in the GH/IGF-1 vocabulary — somatotroph, secretagogue, IGFBP, acid-labile subunit — are defined in the peptide glossary. None of this handling detail changes the pharmacology; meticulous technique preserves whatever activity the molecule has but cannot alter the scope of what has been demonstrated about it.
The measurement point is more scientifically consequential. Because the entire mechanistic story terminates in IGF-1, any rigorous study of tesamorelin’s axis engagement lives or dies by the quality of the IGF-1 assay. IGF-1 immunoassays are notoriously method-dependent: results must be interpreted against age- and sex-specific reference ranges, ideally expressed as standard-deviation scores rather than raw concentrations, and are confounded by binding-protein interference, nutritional and hepatic status, and assay standardization differences between platforms. A single spot GH level, by contrast, is nearly uninterpretable given the pulsatile secretion described earlier. This is why the clinical program and the label both anchor monitoring to IGF-1 SDS rather than to GH, and why any research claim about “how much” tesamorelin raises IGF-1 is meaningful only when the assay and reference framework are specified. The broader catalog of GH-axis compounds and how their parameters are organized for reference is available through the site’s dosages index.
Frequently Asked Questions
Does tesamorelin contain or act as growth hormone or IGF-1?
No. Tesamorelin is a stabilized analog of growth-hormone-releasing hormone (GHRH), the hypothalamic peptide that instructs the pituitary to make and release growth hormone.3 It contains no growth hormone and no IGF-1. It acts several steps upstream, at the GHRH receptor on pituitary somatotrophs, prompting the body to secrete its own GH; that endogenous GH then acts on the liver to raise IGF-1.4 This upstream position is the reason its effects are filtered through the pituitary’s own feedback control rather than bypassing it as injected GH would.
By what pathway does tesamorelin raise IGF-1?
Through a defined multi-organ cascade. Tesamorelin activates the pituitary GHRH receptor (a class B GPCR) via Gαs/cAMP/PKA, driving pulsatile GH release.4 The released GH binds hepatic GH receptors, activating JAK2, which phosphorylates STAT5b; STAT5b enters the nucleus and transcribes the IGF1 gene, and the liver secretes IGF-1 into the blood.67 The circulating IGF-1, stabilized on its binding proteins, is the steady elevation measured in trials.2
Is the IGF-1 elevation the goal of treatment?
Not exactly. In the approved use, the goal is reduction of excess visceral abdominal fat in HIV-associated lipodystrophy; the IGF-1 rise is the biomarker showing the GH/IGF-1 axis has been engaged, and it correlates with the visceral-fat response.8 Importantly, IGF-1 is also the safety parameter: the label advises considering discontinuation if IGF-1 rises persistently beyond about 3 standard deviations above normal.1 So the aim is to bring IGF-1 into a therapeutic window under monitoring, not to maximize it.
What is tesamorelin actually FDA-approved for?
One thing only: to reduce excess visceral abdominal fat in adults with HIV-associated lipodystrophy, marketed as Egrifta.1 It is not approved as a general weight-loss drug, an anti-aging therapy, a muscle-building agent, or a treatment for fatty liver disease in the general population. Uses outside the approved indication are off-label or investigational.
Why does tesamorelin preserve pulsatile GH secretion when injected GH does not?
Because it works by prompting the pituitary to release its own GH rather than infusing hormone directly. The somatotroph remains under the control of hypothalamic somatostatin and of negative feedback from GH and IGF-1, so tesamorelin amplifies the existing rhythmic pulses instead of imposing a flat, continuous level.35 Target tissues read the pattern of GH exposure, and pulsatility allows receptors to reset between peaks.5 This is a mechanistic distinction, not a proven claim of clinical superiority over recombinant GH.
How large and how durable is the IGF-1 increase?
In the pivotal HIV-lipodystrophy trials, once-daily tesamorelin 2 mg produced a clear IGF-1 rise versus a nearly flat placebo response, and the increase was sustained through roughly a year of continuous dosing without tachyphylaxis.28 The exact magnitude varies between individuals because IGF-1 output depends on hepatic health, insulin and nutritional status, and STAT5b-pathway integrity, and because IGF-1 assays are method-dependent. Stopping the drug allows IGF-1 to fall back toward baseline.8
What are the main risks tied to the IGF-1 elevation?
Because IGF-1 is a growth and survival factor, tesamorelin is contraindicated in active malignancy, and its effect on the development or progression of cancer is unknown, so IGF-1 must be monitored.1 GH-axis amplification can also impair glucose tolerance and cause fluid retention, arthralgia, edema, and carpal-tunnel-type symptoms.11 These are the predictable consequences of activating the axis, and they are why the approved use is confined to a monitored population.12
Does raising IGF-1 with tesamorelin build muscle or reverse aging?
That is not what the approved evidence shows. The registrational trials measured visceral fat, lipids, glucose, and IGF-1 in HIV-associated lipodystrophy — not muscle strength, physical function, or aging outcomes.28 While the GH/IGF-1 axis is biologically involved in tissue anabolism, extrapolating from “tesamorelin raises IGF-1” to “tesamorelin builds muscle or slows aging” is an inference the human data do not support, and such uses are investigational at best.
References
- EGRIFTA SV / EGRIFTA WR (tesamorelin) full prescribing information. Theratechnologies; U.S. Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/022505s020lbl.pdf
- Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359-2370. PMID: 18057338. https://pubmed.ncbi.nlm.nih.gov/18057338/
- Dhillon S. Spotlight on tesamorelin in HIV-associated lipodystrophy. BioDrugs. 2011;25(6):405-408. PMID: 22050344. https://pubmed.ncbi.nlm.nih.gov/22050344/
- Halmos G, Szabo Z, Dobos N, Juhasz E, Schally AV. Growth hormone-releasing hormone receptor (GHRH-R) and its signaling. Rev Endocr Metab Disord. 2025. PMCID: PMC12137518. https://pmc.ncbi.nlm.nih.gov/articles/PMC12137518/
- Normal Physiology of Growth Hormone in Normal Adults. In: Endotext [Internet]. South Dartmouth (MA): MDText.com. Bookshelf ID: NBK279056. https://www.ncbi.nlm.nih.gov/books/NBK279056/
- Dehkhoda F, Lee CMM, Medina J, Brooks AJ. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front Endocrinol (Lausanne). 2018;9:35. PMID: 29487568. https://pubmed.ncbi.nlm.nih.gov/29487568/
- Chia DJ. Minireview: mechanisms of growth hormone-mediated gene regulation. Mol Endocrinol. 2014;28(7):1012-1025. PMID: 24825400. https://pubmed.ncbi.nlm.nih.gov/24825400/
- Falutz J, Mamputu JC, Potvin D, et al. Effects of tesamorelin (TH9507), a growth hormone-releasing factor analog, in HIV-infected patients with excess abdominal fat: a pooled analysis of two multicenter, double-blind placebo-controlled phase 3 trials with safety extension data. J Clin Endocrinol Metab. 2010;95(9):4291-4304. PMID: 20554713. https://pubmed.ncbi.nlm.nih.gov/20554713/
- Stanley TL, Feldpausch MN, Oh J, et al. Effect of tesamorelin on visceral fat and liver fat in HIV-infected patients with abdominal fat accumulation: a randomized clinical trial. JAMA. 2014;312(4):380-389. PMID: 25038357. https://pubmed.ncbi.nlm.nih.gov/25038357/
- Stanley TL, Fourman LT, Feldpausch MN, et al. Effects of tesamorelin on non-alcoholic fatty liver disease in HIV: a randomised, double-blind, multicentre trial. Lancet HIV. 2019;6(12):e821-e830. PMID: 31611038. https://pubmed.ncbi.nlm.nih.gov/31611038/
- Tesamorelin. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. National Institute of Diabetes and Digestive and Kidney Diseases. Bookshelf ID: NBK548730. https://www.ncbi.nlm.nih.gov/books/NBK548730/
- Body composition, hepatic fat, metabolic, and safety outcomes of tesamorelin, a GHRH analogue, in HIV-associated lipodystrophy: a meta-analysis of randomized controlled trials. Obes Res Clin Pract. 2026. PMID: 41545261. https://pubmed.ncbi.nlm.nih.gov/41545261/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. Tesamorelin (Egrifta) is approved by the U.S. Food and Drug Administration only to reduce excess visceral abdominal fat in adults with HIV-associated lipodystrophy; it is not approved for weight loss in the general population, anti-aging, muscle building, cognitive enhancement, or the treatment of non-alcoholic fatty liver disease, and any such use is off-label or investigational. The IGF-1 elevation described here is a real, monitored biomarker of growth-hormone-axis engagement, not a therapeutic endpoint to be maximized. Nothing here is medical advice or a recommendation for human use. Tesamorelin activates the growth hormone/IGF-1 axis and carries specific risks, including effects on glucose tolerance and unknown effects on malignancy; it is a prescription medication that requires clinical supervision and IGF-1 monitoring. Readers should consult qualified healthcare professionals and applicable regulations before making any decisions.