The question in this article’s title carries a quiet assumption that is worth exposing before we accept it. To ask whether NAD+ deficiency is “implicated in” neurodegenerative disease progression is to hover between two very different claims. The weak version merely says that NAD+ levels fall alongside neurodegeneration — an observation that is well documented and not seriously disputed. The strong version says that the fall in NAD+ is a mechanistic driver of disease progression, a causal lever whose collapse pushes neurons toward death and whose restoration might slow the disease. Those are not the same statement, and most of the confident language circulating online quietly slides from the first to the second as if the gap did not exist.
This piece treats the title as an open research question rather than a settled fact, because that is what the evidence supports. NAD+ — nicotinamide adenine dinucleotide — is a coenzyme found in every living cell, and its two supplemental precursors most discussed in this context, nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), are marketed as dietary supplements. Neither NAD+ itself nor its precursors are approved by the U.S. Food and Drug Administration, the European Medicines Agency, or any comparable regulator to treat, prevent, or slow Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease, or any other neurodegenerative condition. The great majority of the mechanistic evidence linking NAD+ loss to neurodegeneration comes from cell culture and rodent models. Human evidence exists, but it is thin: a handful of small, mostly phase I trials that establish safety and biomarker changes, with clinical signals that are best described as preliminary and exploratory.89
So the honest map looks like this. There is a strong and biologically coherent hypothesis that NAD+ decline participates in the machinery of neurodegeneration; there is substantial preclinical support for that hypothesis; there is a real and unresolved question of whether the decline is a cause, a consequence, or both; and there is very little human outcome data to tell us whether raising NAD+ changes the course of any of these diseases. This article walks through each of those layers — what NAD+ does in the brain, the evidence that it declines, the mechanistic pathways proposed to connect that decline to neuronal loss, the peculiar and instructive case of axonal degeneration, the chicken-and-egg problem of causation, what the human trials actually show, and the regulatory and safety reality — while refusing at every step to inflate a plausible mechanism into a proven therapy.
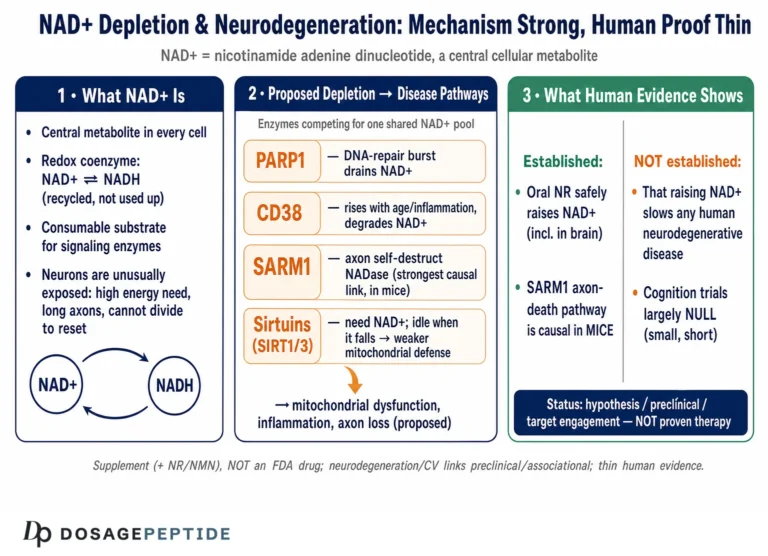
What NAD+ Is and Why Neurons Are Especially Exposed
NAD+ is a small molecule that wears two hats. In its first and oldest role it is a redox coenzyme: it accepts and donates electrons, cycling between the oxidized form (NAD+) and the reduced form (NADH), and in doing so it is indispensable to glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation — the reactions that generate cellular ATP.1 In this capacity NAD+ is not consumed; it is recycled, shuttling electrons back and forth thousands of times.
Its second hat is what makes it interesting for disease biology. NAD+ is also a cosubstrate that is physically consumed by three families of enzymes: the sirtuins (SIRT1–7), the poly(ADP-ribose) polymerases (PARPs), and the NAD glycohydrolases CD38 and the more recently characterized SARM1.1 When these enzymes act, they cleave NAD+ and use its nicotinamide-and-ADP-ribose components as building blocks for signaling reactions — deacetylating proteins, tagging damaged DNA with poly(ADP-ribose), generating calcium-mobilizing second messengers. Every one of these reactions burns a molecule of NAD+ that must then be resynthesized. The cell’s NAD+ level at any moment is therefore a running balance between synthesis (chiefly the salvage pathway, in which the enzyme NAMPT recycles nicotinamide back into the pool) and consumption by these enzyme families.111
Neurons sit at an uncomfortable intersection of this economy. They are among the most energy-hungry cells in the body, spending enormous amounts of ATP to maintain ion gradients and fire action potentials, which keeps redox demand for NAD+/NADH constantly high. They are post-mitotic and long-lived, so they cannot dilute accumulated damage by dividing and must instead repair it in place — a process that leans heavily on the NAD+-consuming PARP enzymes. And their extraordinarily long axons pose a distribution problem: NAD+ and the machinery that makes it must be trafficked far from the cell body, leaving distal axons vulnerable if that supply falters. Each of these features means that a neuron is unusually sensitive to any disturbance in the NAD+ balance, and it is this sensitivity, more than any single pathway, that makes the NAD+ hypothesis of neurodegeneration biologically plausible in the first place.10
It is worth being precise about terminology, because sloppy usage drives much of the confusion. “NAD+” strictly denotes the oxidized coenzyme; “NAD” or “total NAD” often refers to the combined pool of NAD+ and NADH; and the “NAD metabolome” encompasses precursors and intermediates such as nicotinamide, NR, NMN, and the phosphorylated NADP+/NADPH pair. When a study reports that “NAD declines,” it matters enormously whether it measured the oxidized species, the total pool, or the redox ratio, and in which cellular compartment — nucleus, cytosol, or mitochondrion — since these can move independently. Readers new to these distinctions may find the site’s peptide and compound glossary a useful reference for the vocabulary that recurs throughout the literature.
The Evidence That NAD+ Declines — and What Kind of Evidence It Is
The first plank of the hypothesis is empirical: does NAD+ actually fall with aging and in neurodegenerative disease? For aging, the answer in multiple tissues is reasonably solid. In the human brain specifically, a landmark 2015 study used phosphorus-31 magnetic resonance spectroscopy (31P-MRS) at high field to measure intracellular NAD+ and NADH non-invasively, and found that total NAD content and the NAD+/NADH redox ratio decreased with age across healthy adults, while NADH rose.2 This was an important result because it moved the age-related decline from something inferred in rodents and post-mortem tissue to something measured in living human brains.
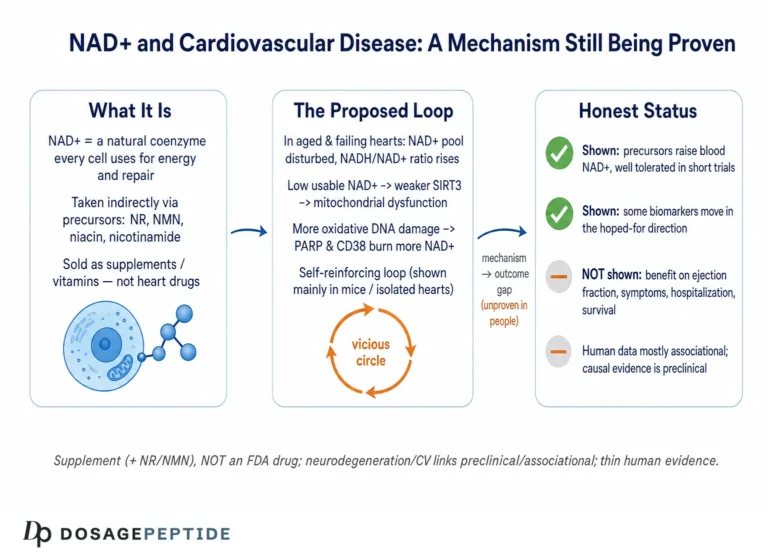
The mechanism behind the age-related decline appears to be as much about accelerated consumption as about failing synthesis. Work in mice identified the NADase CD38 — an enzyme whose expression and activity climb with age, largely in immune and glial cells — as a principal driver of the age-related NAD+ collapse, acting through a mechanism that also compromises the mitochondrial sirtuin SIRT3.3 In parallel, chronic low-grade inflammation and DNA damage activate CD38 and PARP respectively, both of which draw down the shared NAD+ pool. The picture that emerges is of an aging cell facing rising NAD+ demand from consuming enzymes at exactly the time its salvage capacity is under strain.11
For neurodegenerative disease, the evidence is more heterogeneous and, crucially, more associational. Post-mortem and model-system studies report disturbed NAD+ metabolism in Alzheimer’s disease, Parkinson’s disease, and other conditions — altered levels of NAD+ precursors, changes in NAMPT expression, elevated markers of PARP activation, and shifts in the enzymes that regulate the pool.1112 But most of these are cross-sectional snapshots or come from animal models, and a cross-sectional association cannot by itself tell us whether low NAD+ preceded the disease, resulted from it, or simply travelled alongside it. This is the central interpretive limitation to keep in mind: an abundance of studies showing that NAD+ is low in disease does not, on its own, establish that low NAD+ drives the disease. That distinction is the hinge on which the entire question turns, and we return to it in a dedicated section below.
A further caution concerns measurement. Reliable in-vivo NAD+ quantification in the human brain is technically demanding and available at only a handful of centers; much of what we “know” about NAD+ in specific diseases still rests on tissue that is difficult to obtain, on peripheral surrogates such as blood metabolites that may not reflect brain levels, or on animal tissue. The confident statements one encounters that a particular disease “is characterized by NAD+ deficiency” often outrun the granularity of the underlying measurements.
The Proposed Mechanisms: How NAD+ Loss Could Drive Neuronal Death
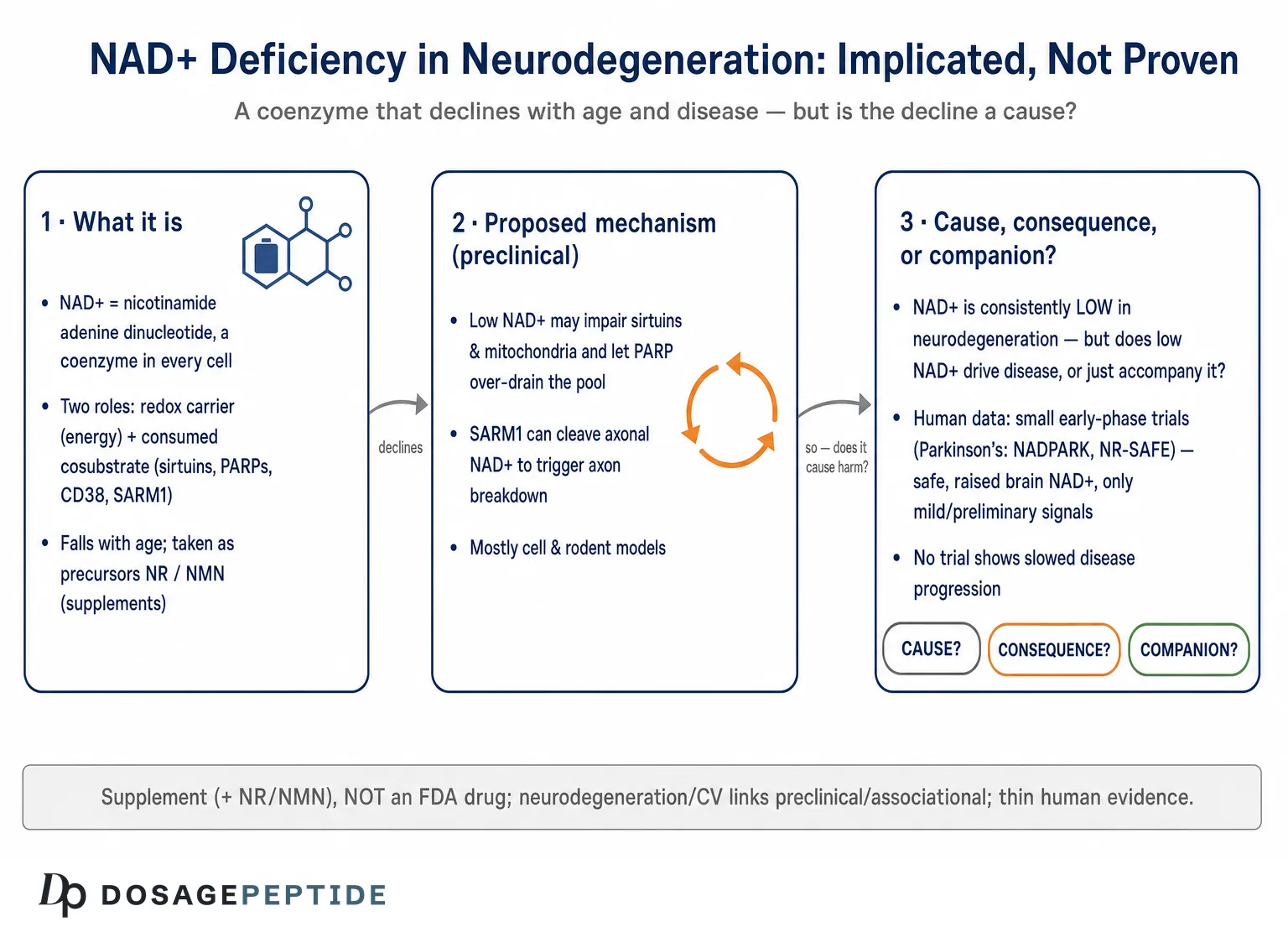
If NAD+ decline does contribute to neurodegeneration, through what machinery would it act? The mechanistic case is not one pathway but a web of interlocking ones, and its internal coherence is part of why the hypothesis is taken seriously. The dominant threads are these.
Sirtuin failure. The sirtuins are NAD+-dependent deacetylases that regulate mitochondrial biogenesis, antioxidant defenses, inflammation, and (in the case of SIRT1 and SIRT6) genomic stability. Because their activity is gated by NAD+ availability, a falling NAD+ pool directly throttles sirtuin function. SIRT1 in neurons and SIRT3 in mitochondria have both been implicated in protective programs, and their impairment under NAD+ scarcity is proposed to remove a brake on oxidative and inflammatory damage.111 This is the pathway most often invoked to connect NAD+ to the “healthy aging” literature, and it is genuinely well supported at the biochemical level — though establishing that sirtuin loss is rate-limiting for a human disease is another matter.
PARP hyperactivation. Poly(ADP-ribose) polymerase-1 (PARP-1) is activated by DNA strand breaks and consumes large quantities of NAD+ to synthesize poly(ADP-ribose) chains as part of the repair response. In neurodegeneration, chronic DNA damage — from oxidative stress, from impaired repair, from aggregated proteins — can drive PARP-1 into sustained overactivity, depleting NAD+ so severely that the cell’s energy metabolism collapses, a form of programmed death sometimes called parthanatos.1112 This creates a vicious cycle: damage activates PARP, PARP drains NAD+, NAD+ depletion impairs the sirtuins and mitochondria that would otherwise limit further damage. This feed-forward loop is one of the more compelling mechanistic arguments that NAD+ is not merely a passive marker but an active participant.
Mitochondrial dysfunction and impaired mitophagy. Because NAD+/NADH is central to oxidative phosphorylation, a depleted or unfavorably shifted pool impairs ATP production precisely in cells that can least afford it. Compounding this, NAD+-dependent processes govern mitophagy — the selective clearance of damaged mitochondria — so NAD+ loss both starves the neuron of energy and lets defective, ROS-leaking mitochondria accumulate. In Alzheimer’s-model neurons, restoring NAD+ with NR was reported to induce mitophagy and improve mitochondrial quality alongside cognitive measures.6
DNA repair capacity. Beyond PARP, the broader DNA-damage response leans on NAD+. In a mouse model engineered to combine Alzheimer’s pathology with a DNA-repair deficiency (3xTgAD crossed with a polymerase-β haploinsufficiency), the cerebral NAD+/NADH ratio was reduced, and NR supplementation normalized it while reducing DNA damage, neuroinflammation, and hippocampal neuronal apoptosis and increasing brain SIRT3 activity.5 This is one of the cleaner demonstrations that manipulating NAD+ moves multiple disease-relevant readouts in the same direction — in a mouse.
Neuroinflammation via CD38 and cGAS–STING. Glial cells are a major node. Pro-inflammatory activation raises CD38 in astrocytes and microglia, which degrades NAD+ and propagates a state of local depletion; conversely, NAD+ repletion has been reported to dampen neuroinflammatory signaling. In an APP/PS1 Alzheimer’s model, NR reduced neuroinflammation and cellular senescence through the cGAS–STING pathway, linking NAD+ status to the innate-immune signaling increasingly implicated in neurodegeneration.6 The genetic counterpart is striking: deleting CD38 in an Alzheimer’s mouse model reduced amyloid-β plaque load and soluble Aβ and improved spatial learning, tying the NAD-consuming enzyme directly to pathology.4
The following table summarizes the principal NAD+-handling enzymes and the direction in which each is thought to push neuronal fate.
| Enzyme / family | Action on NAD+ | Proposed role in neurodegeneration |
|---|---|---|
| Sirtuins (SIRT1, SIRT3) | Consume NAD+ to deacetylate targets | Protective; activity falls when NAD+ is scarce, removing a brake on damage1 |
| PARP-1 | Consume NAD+ to poly(ADP-ribosyl)ate at DNA breaks | Repair when moderate; NAD+-depleting and lethal when hyperactivated11 |
| CD38 | NAD glycohydrolase; degrades NAD+/NMN | Rises with age and inflammation in glia; drives NAD+ decline; deletion is protective in AD models34 |
| SARM1 | TIR-domain NADase, latent until activated | Executioner of axon degeneration; cleaves axonal NAD+ to trigger fragmentation7 |
| NAMPT (synthetic) | Rate-limiting salvage enzyme, regenerates NAD+ | Supports the pool; reduced expression with inflammation lowers NAD+1 |
Two honest observations about this mechanistic web. First, it is genuinely coherent: the pathways reinforce one another, and interventions that raise NAD+ tend to move several of them favorably at once in model systems. That coherence is a real point in the hypothesis’s favor. Second, coherence is not proof. A mechanism that makes sense on a whiteboard and works in a mouse has, historically, a discouraging track record of translating into disease-modifying human therapy for neurodegeneration — a field littered with mechanistically elegant failures. The mechanistic case tells us the hypothesis is worth testing; it does not tell us the hypothesis is true in humans.
The Axonal Case: SARM1 and NAD+ as Both Fuel and Fuse
One corner of this field deserves separate treatment because it is where the causal role of NAD+ metabolism is most firmly established — and where it complicates the naive “more NAD+ is better” framing in an instructive way. That corner is programmed axon degeneration.
Axons maintain their integrity in part through a constant supply of the labile survival factor NMNAT2, an enzyme that synthesizes NAD+ locally. When an axon is injured, or when NMNAT2 supply fails in disease, the guardian enzyme SARM1 — normally held in an auto-inhibited state — is unleashed. SARM1 is itself an NADase: once activated, it catastrophically cleaves axonal NAD+, and this depletion executes the fragmentation of the axon (the process classically observed as Wallerian degeneration after nerve injury).7 Critically, SARM1 is switched on not by absolute NAD+ levels alone but by a rising ratio of NMN to NAD+: as NMNAT2 is lost, its substrate NMN accumulates while its product NAD+ falls, and this shift in the NMN/NAD+ ratio is the trigger that disinhibits SARM1.7
This matters for the article’s central question in two ways. First, it is a bona fide example where NAD+ metabolism is not a bystander but the actual execution mechanism of neuronal (axonal) destruction, with a clear causal chain established in animals and increasingly implicated in peripheral neuropathies and possibly in ALS and other axonopathies. Genetic loss of SARM1 protects axons across numerous injury and disease models, which is about as strong a causal statement as this field offers.7
Second, and this is the twist that should make anyone cautious about simplistic supplementation logic: because SARM1 is triggered by the NMN/NAD+ ratio, giving the precursor NMN can in some contexts promote axon degeneration by raising NMN faster than it is converted to NAD+, whereas strategies that boost NAD+ directly, or that lower NMN, are protective.7 In other words, within this one well-characterized system, the two most popular NAD+ precursors are not interchangeable, and one of them can theoretically do harm. This is a powerful reminder that “NAD+ deficiency” is not a single dial you turn up with any precursor; the intermediates matter, the ratios matter, and the same molecule can protect or damage depending on the enzymatic context. Anyone reasoning from “NAD+ is low in neurodegeneration” straight to “therefore take an NAD+ precursor” is skipping over exactly the kind of nuance the SARM1 story exposes.
Cause, Consequence, or Companion? The Central Unresolved Problem
We now reach the question the title implicitly asks and that most popular writing dodges. When NAD+ is found to be low in a neurodegenerating brain, which of the following is true: (a) the NAD+ deficit is a cause that accelerates disease; (b) it is a consequence of disease processes that consume or fail to synthesize NAD+; or (c) it is a companion, co-varying with disease without a strong causal link in either direction? The uncomfortable but honest answer is that in most neurodegenerative diseases we do not yet know, and the three possibilities are not mutually exclusive.
The arguments that the deficit is at least partly causal are the mechanistic ones already surveyed: the PARP–NAD+–sirtuin feed-forward loop, the SARM1 axon-execution pathway, and the consistent finding that restoring NAD+ in animal models improves pathology and behavior.567 The intervention studies are the strongest form of this argument, because manipulating the putative cause and observing an effect is closer to a causal test than simply observing a correlation. If lowering NAD+ worsens outcomes and raising it improves them within the same model, causation becomes hard to dismiss — at least in that model.
The arguments that the deficit is substantially a consequence are equally real. Neurodegeneration generates enormous DNA damage and neuroinflammation, both of which activate NAD+-consuming enzymes (PARP, CD38); mitochondrial failure impairs the energy metabolism that maintains the NAD+/NADH balance; and dying cells simply lose their metabolic machinery. On this reading, low NAD+ is a downstream readout of upstream pathology — a smoke detector, not the fire. If so, boosting NAD+ might treat a symptom of the disturbance without touching its root, which could explain why biomarker improvements in animals might not translate into disease modification in humans.
Distinguishing these possibilities requires evidence that observational and even most interventional animal studies cannot fully provide. What would help are: longitudinal human data showing that NAD+ decline precedes clinical or pathological progression rather than accompanying it; Mendelian-randomization-style genetic evidence that variants affecting NAD+ metabolism alter disease risk; and, decisively, adequately powered, long-duration randomized controlled trials in which raising NAD+ changes hard clinical outcomes. Very little of this exists. The animal interventions, however elegant, are performed in models that reproduce only fragments of the human diseases, over short timescales, in genetically uniform animals — conditions under which many interventions “work” that later fail in people. Readers exploring how this cause-versus-consequence problem plays out in a single disease may find the companion analysis of the link between NAD+ deficiency and Parkinson’s progression a useful, more focused case study of the same interpretive tension.
A useful way to hold this ambiguity is to notice that the same evidence can be read as supporting either interpretation depending on which end of the causal arrow one privileges. Consider the observation that NAD+ repletion improves outcomes in an Alzheimer’s mouse: a causation advocate reads this as “correcting the deficit fixes the disease,” while a consequence advocate reads it as “a metabolic booster provides symptomatic support to stressed neurons without touching the amyloid or tau pathology that defines the disease.” Both readings are compatible with the data, and the mouse experiment, however clean, cannot adjudicate between them because it does not follow the animals to a hard endpoint under conditions that mirror decades of human disease. This is not a rhetorical stalemate; it is a genuine limit of what the current experimental designs can establish, and recognizing it is the difference between an expert’s reading and a marketer’s.
The intellectually honest position, then, is that NAD+ deficiency is implicated in neurodegenerative progression in the weak-to-moderate sense — it is consistently present, mechanistically connected, and modifiable in models — but that its status as a primary driver whose correction would slow human disease remains an open question. The title’s premise is a live hypothesis under active investigation, not an established mechanism.
What the Human Trials Actually Show
Because this is where hope most often outruns data, it is worth laying out the human clinical record precisely. As of mid-2026, the controlled human evidence for NAD+ precursors in neurodegenerative disease consists mainly of small, short, early-phase trials focused on safety, brain bioavailability, and biomarkers — not on demonstrating that disease progression is slowed.
The most informative example is Parkinson’s disease. The NADPARK study, published in Cell Metabolism in 2022, was a double-blind, randomized, placebo-controlled phase I trial in which 30 newly diagnosed, treatment-naive patients received 1,000 mg of nicotinamide riboside or placebo for 30 days.8 Its findings were carefully framed by the investigators themselves: NR was well tolerated and produced a significant but variable increase in cerebral NAD levels (measured by 31P-MRS) and related cerebrospinal-fluid metabolites; the subset of recipients who showed a clear brain-NAD increase exhibited altered cerebral metabolism on FDG-PET, and this was associated with a mild clinical improvement.8 The appropriate reading is that NR can raise brain NAD+ in some people and that this correlates with metabolic and modest symptomatic changes over a month — a promising signal generating hypotheses, not evidence of disease modification.
The follow-up NR-SAFE trial, published in Nature Communications in 2023, was a randomized, double-blind, placebo-controlled phase I safety study of high-dose NR (3,000 mg per day) for four weeks in 20 patients with Parkinson’s disease.9 Its primary purpose was tolerability: it found that high-dose NR was safe, robustly augmented the systemic NAD metabolome, and did not deplete methyl donors, supporting the use of doses up to 3,000 mg per day in future phase 2 trials with monitoring.9 A symptomatic signal was again reported, but the study was neither designed nor powered to demonstrate a clinical benefit, and its authors present it as a safety and dose-ranging step. Larger dose-optimization trials (the N-DOSE program) have followed, and the field’s explicit posture is that phase 2 efficacy testing is the next step — which is another way of saying efficacy has not been established.
For Alzheimer’s disease, the human record is thinner still. Despite a robust preclinical literature, well-controlled clinical trials of NAD+ precursors with cognitive or biomarker endpoints are only in early phases; dose-optimization and bioavailability studies (such as the N-DOSE AD program) are underway, and no large trial has demonstrated that NR or NMN slows cognitive decline or alters Alzheimer’s pathology in patients.1213 The gap between the density of the mouse literature and the sparseness of the human outcome data is itself one of the most important facts in this entire area.
| Trial / program | Population & design | Intervention | What it showed |
|---|---|---|---|
| NADPARK (2022) | 30 treatment-naive PD patients; RCT, phase I, 30 days8 | NR 1,000 mg/day vs placebo | Safe; variable rise in brain NAD; altered cerebral metabolism and mild clinical improvement in responders |
| NR-SAFE (2023) | 20 PD patients; RCT, phase I safety, 4 weeks9 | NR 3,000 mg/day vs placebo | Safe at high dose; strong systemic NAD augmentation; no methyl-donor depletion; symptomatic signal, not powered for efficacy |
| N-DOSE / N-DOSE AD | Dose-optimization in PD and AD; early phase12 | NR, escalating doses | Ongoing; aims to define optimal dose and cerebral bioavailability — efficacy not yet established |
| AD preclinical body | Cell and mouse models (3xTgAD/Polβ, APP/PS1)56 | NR / NAD+ repletion | Reduced DNA damage, neuroinflammation, senescence; improved mitophagy and cognition — in animals only |
The pattern across the table is unmistakable and should govern expectations: robust preclinical benefit, promising early-phase human safety and biomarker data, and an absence of the large, long, outcome-focused randomized trials that would be needed to call NAD+ repletion a disease-modifying therapy. For a broader view of how NAD+ has been studied in humans beyond neurodegeneration — including cellular-repair and longevity endpoints — the discussion of how NAD+ influences cellular damage repair and longevity in human studies surveys the same evidentiary caution in an adjacent context.
Disease by Disease: Where the Evidence Is Strongest and Weakest
Neurodegeneration is not one disease, and the NAD+ hypothesis stands on different footing in each. Collapsing them into a single “neurodegenerative disease” category — as the article title does, and as much marketing does — obscures that the quality of evidence varies markedly.
Parkinson’s disease currently has the strongest human NAD+ evidence, precisely because the NADPARK and NR-SAFE trials were done there. Mitochondrial dysfunction is a well-established feature of PD, which gives the NAD+ energy-metabolism rationale a natural home, and the trials show that brain NAD+ can be augmented with a plausible metabolic response.89 But even here, the clinical signals are mild and preliminary, and no trial has yet shown slowed progression.
Alzheimer’s disease has the richest preclinical NAD+ literature — the DNA-repair, mitophagy, neuroinflammation, and CD38 studies are among the most detailed mechanistic work in the field.456 Yet the human clinical evidence lags far behind, consisting of early bioavailability and dose studies rather than efficacy trials.12 The AD story is thus a cautionary illustration of the preclinical-to-clinical gap.
Amyotrophic lateral sclerosis and peripheral neuropathies connect most directly to the SARM1 axon-degeneration mechanism, which is arguably the best-established causal link between NAD+ metabolism and neuronal loss.7 This has driven intense interest in SARM1 inhibitors as a therapeutic class — a strategy conceptually distinct from simply supplementing NAD+ precursors, and one still in preclinical or early clinical development.
Huntington’s disease and other conditions have supportive mechanistic and model-based associations with NAD+ metabolism, but comparatively little disease-specific human data, and should be regarded as areas of hypothesis rather than evidence.11
| Disease | Mechanistic rationale | Human evidence level |
|---|---|---|
| Parkinson’s disease | Mitochondrial dysfunction; energy-metabolism deficit | Small phase I RCTs (NADPARK, NR-SAFE); mild signals; no proven disease modification89 |
| Alzheimer’s disease | DNA repair, mitophagy, neuroinflammation, CD38 | Early bioavailability/dose studies only; no efficacy trials12 |
| ALS / neuropathy | SARM1-driven axon degeneration (NMN/NAD+ ratio) | Strong preclinical causal link; SARM1-inhibitor programs early7 |
| Huntington’s / other | Sirtuin, PARP, mitochondrial associations | Largely preclinical/associational; minimal human data11 |
The disaggregated view corrects a common error. Someone who has read that “NAD+ helps Parkinson’s” and someone who has read that “NAD+ reverses Alzheimer’s in mice” may both conclude that “NAD+ treats neurodegeneration,” but the first is a modest human signal and the second is a mouse result, and merging them into one confident claim is exactly the kind of evidence inflation this article exists to resist.
NAD+ Precursors, Supplements, and the Regulatory Reality
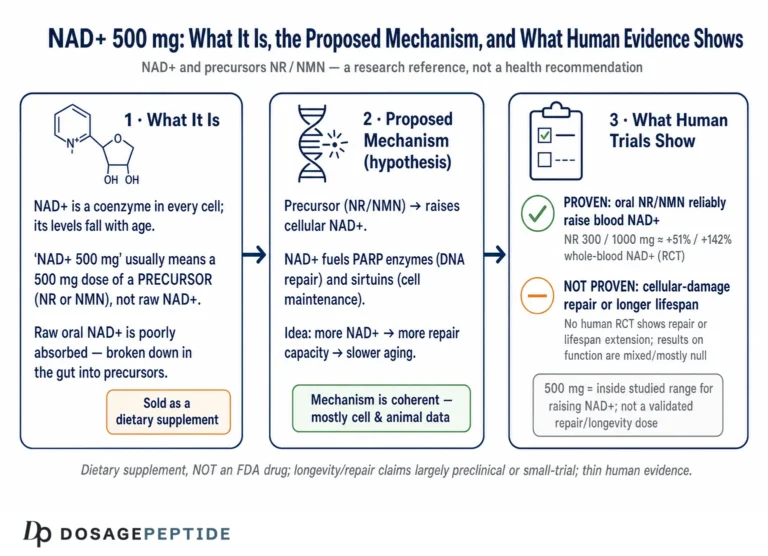
It is essential to be clear about what NAD+ and its precursors are in a legal and regulatory sense, because this shapes both safety and the interpretation of claims. NAD+ itself is not typically supplemented directly with good oral bioavailability; instead, people take precursors. Nicotinamide (niacinamide) and nicotinic acid are forms of vitamin B3. Nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are newer precursors marketed as dietary supplements.
In the United States, NR is sold as a dietary-supplement ingredient. NMN has had a more contested regulatory history: the FDA has taken the position that NMN is excluded from the dietary-supplement definition because it has been authorized for investigation as a new drug, a stance that has generated ongoing industry dispute and affects its legal marketing. In no case is any NAD+ precursor approved as a drug to treat, prevent, or slow any neurodegenerative disease. Dietary supplements, by law, may not be marketed with claims to diagnose, treat, cure, or prevent disease; claims that a product “treats Parkinson’s” or “prevents Alzheimer’s” are outside what the evidence and the regulations permit.
Several practical consequences follow. First, supplement-grade products are not held to the manufacturing, purity, and dose-verification standards of approved pharmaceuticals; content can vary between labels and lots. Second, the doses used in the informative clinical trials (for example, 1,000–3,000 mg/day of NR under medical supervision with monitoring) were studied over weeks, not the years that disease modification would require; long-term safety at high doses is not fully characterized.89 Third, as the SARM1 story showed, precursors are not biochemically interchangeable, and the assumption that any “NAD+ booster” is equivalent to any other is unfounded.7
On safety, the reassuring finding from the trials is that NR, even at 3,000 mg/day for four weeks, was well tolerated with only mild adverse events and no methyl-donor depletion in the studied population.9 That is a genuine and useful result. But short-term tolerability in small trials of relatively selected patients is not the same as long-term safety in the broad, often elderly and polypharmacy-burdened populations who live with neurodegenerative disease, and it is certainly not evidence of benefit. Absence of demonstrated harm and absence of demonstrated efficacy can coexist — and here, for disease modification, they do. Those seeking the general vocabulary and handling conventions that recur in this literature can consult the site’s reconstitution and handling guide and its central dosages reference, both of which are organized for educational purposes rather than as guidance for human treatment.
Why the Mechanism Is Attractive but Translation Is Hard
It is worth pausing on why the NAD+ hypothesis has attracted so much attention and investment, and why that enthusiasm warrants tempering. The appeal is real and multi-layered. NAD+ sits at the convergence of aging, metabolism, DNA repair, inflammation, and mitochondrial function — each independently implicated in neurodegeneration — so a single molecule appears to offer a unifying node. It is measurable, at least in principle, and modifiable with oral precursors that are inexpensive and appear safe short-term. And the aging connection lends it narrative power, since neurodegenerative diseases are overwhelmingly age-associated and NAD+ demonstrably falls with age.12 A modifiable, measurable, mechanistically central, aging-linked target is close to an ideal that drug developers dream of.
The reasons for caution are equally structural. Neurodegenerative diseases develop over decades, so any truly disease-modifying intervention likely needs to be started early and sustained for years — a scenario that short trials cannot test and that raises the stakes for long-term safety. The blood–brain barrier and the question of how much oral precursor actually reaches and is used by the relevant neurons remain incompletely resolved; NADPARK’s finding that the brain-NAD response to NR was variable across individuals is a direct signal of this problem.8 The redundancy and compartmentalization of NAD+ metabolism mean that raising a whole-brain or blood NAD+ measure may not correct the specific compartmental deficit that matters in a given cell. And, as emphasized throughout, correcting a downstream metabolic marker does not necessarily touch upstream disease drivers such as protein aggregation.
There is also a subtler translational trap worth naming. In young, genetically homogeneous rodents, an intervention often works because the modeled deficit is the dominant problem the animal faces; the model is, in effect, engineered so that NAD+ metabolism is rate-limiting. Human neurodegenerative disease is not engineered that way. It arrives layered on top of decades of vascular change, mixed pathologies (many older patients have simultaneous amyloid, tau, vascular, and synuclein burdens), variable genetics, and a dozen comorbidities. In that noisy setting, correcting one node — even a genuinely important one — may produce an effect too small to detect against the background, or may be swamped by pathologies the intervention never addresses. The very feature that makes a mouse study interpretable, its isolation of a single variable, is what makes its result a fragile predictor of the human context. This is the general reason a mechanistically central, model-validated target can still disappoint in trials, and NAD+ is not exempt from it.
The field’s own framing reflects this tension. Recent expert reviews describe NAD+ augmentation as a candidate disease-modifying strategy — language that correctly signals promise-with-uncertainty rather than established therapy.13 That is the register in which any responsible discussion of the title’s question should be conducted. Comparable caution applies to related growth-factor approaches to age-related cognitive decline; the parallel discussion of whether sermorelin supports cognitive function in age-related neurodegeneration illustrates how easily a plausible mechanism can be mistaken for a proven one across the peptide and metabolic-compound literature.
Limitations and Open Questions
Drawing the threads together, the limitations that bear on the title’s question are substantial and, importantly, they compound one another rather than sitting in isolation.
Causal direction is unresolved. The single largest limitation is that, for most neurodegenerative diseases, we cannot yet say whether NAD+ deficiency is a driver, a consequence, or a companion of progression. The mechanistic and animal-intervention evidence supports a causal contribution, but the definitive human evidence — longitudinal, genetic, and large randomized outcome data — is largely absent.
The preclinical-to-clinical gap is wide. The mouse and cell literature is rich and consistent; the human outcome literature is sparse and preliminary. Neurodegeneration is a field where this gap has repeatedly proven treacherous, with numerous mechanistically sound interventions failing in patients after succeeding in models.5612
Measurement is hard. Reliable brain NAD+ quantification in living humans is available at few centers, and peripheral surrogates may not reflect the compartment-specific, cell-type-specific deficits that matter. Many disease-specific claims rest on measurements coarser than the claims themselves.2
Precursors are not interchangeable, and more is not simply better. The SARM1 example shows that the same intervention logic can protect or harm depending on which intermediate is raised and in what ratio — a decisive caution against treating “NAD+ boosting” as a single, uniformly beneficial action.7
Regulatory and quality realities constrain inference. Because precursors are supplements rather than approved drugs, real-world products vary in quality, long-term high-dose safety is uncharacterized, and no disease-treatment claim is legally or evidentially supported.
The honest synthesis is that NAD+ deficiency is implicated in neurodegenerative progression in the sense that it is consistently present, mechanistically interwoven with core disease pathways, and modifiable in models — but that whether it is a primary, correctable driver of human disease progression remains a genuine open question, and the leap from “implicated” to “treatable target” is one the current evidence does not license. For readers tracking how these questions evolve, the site’s discussions of the mechanisms linking NAD+ to DNA repair and the evidence on NAD+ and neuronal preservation during stroke examine adjacent facets of the same underlying biology with the same evidentiary caution.
Frequently Asked Questions
Does NAD+ deficiency cause neurodegenerative disease?
The honest answer is that this is not established. NAD+ consistently declines with aging and is found to be disturbed in neurodegenerative disease, and mechanistic and animal studies show that NAD+ loss can drive damage through pathways such as PARP hyperactivation, sirtuin failure, mitochondrial dysfunction, and SARM1-mediated axon degeneration.1711 But most human data are cross-sectional associations, which cannot separate cause from consequence. NAD+ deficiency is best described as implicated in disease progression, not proven to cause it.
Can taking NAD+ or NR/NMN supplements slow or prevent Alzheimer’s or Parkinson’s?
There is no proof that they can. NAD+ precursors are dietary supplements, not approved drugs for any neurodegenerative disease. The most informative human trials, in Parkinson’s disease (NADPARK and NR-SAFE), were small, short, early-phase studies focused on safety and brain NAD+ levels; they reported only mild, preliminary clinical signals and were not designed to show slowed progression.89 No large trial has demonstrated disease modification, and claims that a supplement treats or prevents these diseases are not supported by the evidence.
Why does NAD+ fall with age?
The decline appears to reflect both increased consumption and altered synthesis. The NADase CD38 rises with age and inflammation, degrading NAD+; DNA damage activates PARP, which consumes NAD+; and salvage-pathway capacity is strained.3 In the human brain, total NAD and the NAD+/NADH redox ratio have been shown to decrease with age using 31P-MRS.2
Is NMN the same as NR for the brain?
No — and this matters. In the SARM1 axon-degeneration pathway, degeneration is triggered by a rising NMN/NAD+ ratio, so raising NMN can in some contexts promote axon damage, whereas raising NAD+ directly is protective.7 The two precursors are not biochemically interchangeable, and the assumption that any “NAD+ booster” behaves identically is unfounded.
What does the SARM1 pathway tell us?
SARM1 is the clearest example of NAD+ metabolism acting as a genuine execution mechanism of neuronal (axonal) destruction: when activated, it cleaves axonal NAD+ to trigger fragmentation, and genetically deleting it protects axons across many injury and disease models.7 It is a strong causal link, but it points toward SARM1 inhibition as a strategy distinct from simply supplementing NAD+ precursors.
What is the strongest evidence for the NAD+–neurodegeneration link?
The strongest causal evidence is the SARM1 axon-degeneration mechanism in animals.7 The strongest disease-model evidence is the set of Alzheimer’s mouse studies in which NAD+ repletion reduced DNA damage, neuroinflammation, and senescence and improved mitophagy and cognition.56 The strongest human evidence is the demonstration that NR can raise brain NAD+ in Parkinson’s patients with associated metabolic and mild clinical changes.8 None of these amounts to proof of disease modification in people.
Are NAD+ precursors safe?
In short, controlled trials, NR was well tolerated — even at 3,000 mg/day for four weeks in Parkinson’s patients, with only mild adverse events and no methyl-donor depletion.9 However, short-term tolerability in small trials is not the same as long-term safety in broad, elderly, polypharmacy populations over the years that disease modification would require, and product quality varies because these are supplements rather than regulated drugs.
If NAD+ is low in disease, why isn’t boosting it obviously the answer?
Because low NAD+ may be a consequence rather than a cause, correcting the marker may not touch the underlying disease. Because the deficit is compartment- and cell-type-specific, whole-body or blood measures may not reflect what needs fixing. Because oral precursors reach the brain variably.8 And because, as SARM1 shows, the wrong precursor in the wrong context can theoretically do harm.7 These are precisely the uncertainties that adequately powered human trials are meant to resolve.
Is NAD+ approved by the FDA for neurodegenerative disease?
No. Neither NAD+ nor its precursors NR and NMN are approved by the FDA or comparable regulators to treat, prevent, or slow any neurodegenerative disease. They are marketed as dietary supplements (with NMN’s supplement status contested in the United States), and no disease-treatment claim is legally or evidentially supported.
References
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208-1213. PMID: 26785480. https://pubmed.ncbi.nlm.nih.gov/26785480/
- Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. 2015;112(9):2876-2881. https://www.pnas.org/doi/10.1073/pnas.1417921112
- Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23(6):1127-1139. PMID: 27304511. https://www.cell.com/cell-metabolism/fulltext/S1550-4131(16)30224-8
- Blacher E, Dadali T, Bespalko A, et al. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann Neurol. 2015;78(1):88-103. DOI: 10.1002/ana.24425. https://onlinelibrary.wiley.com/doi/abs/10.1002/ana.24425
- Hou Y, Lautrup S, Cordonnier S, et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci USA. 2018;115(8):E1876-E1885. PMID: 29432159. https://pubmed.ncbi.nlm.nih.gov/29432159/
- Hou Y, Wei Y, Lautrup S, et al. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci USA. 2021;118(37):e2011226118. https://www.pnas.org/doi/10.1073/pnas.2011226118
- Figley MD, DiAntonio A. The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease. Curr Opin Neurobiol. 2020;63:59-66. https://pmc.ncbi.nlm.nih.gov/articles/PMC7483800/
- Brakedal B, Dölle C, Riemer F, et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022;34(3):396-407. PMID: 35235774. https://pubmed.ncbi.nlm.nih.gov/35235774/
- Brakedal B, Tzoulis C, et al. NR-SAFE: a randomized, double-blind safety trial of high dose nicotinamide riboside in Parkinson’s disease. Nat Commun. 2023. https://www.nature.com/articles/s41467-023-43514-6
- Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 2019;30(4):630-655. PMID: 31577933. https://pubmed.ncbi.nlm.nih.gov/31577933/
- Xie N, Zhang L, Gao W, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020;5:227. PMCID: PMC7539288. https://pmc.ncbi.nlm.nih.gov/articles/PMC7539288/
- Yao Z, et al. NAD+ in Alzheimer’s disease: molecular mechanisms and systematic therapeutic evidence obtained in vivo. Front Cell Dev Biol. 2021;9:668491. PMCID: PMC8369418. https://pmc.ncbi.nlm.nih.gov/articles/PMC8369418/
- NAD augmentation as a disease-modifying strategy for neurodegeneration. Trends Endocrinol Metab. 2025. https://www.cell.com/trends/endocrinology-metabolism/fulltext/S1043-2760(25)00070-0
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. NAD+ and its precursors nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are dietary-supplement ingredients, not drugs approved by the FDA, EMA, or any comparable regulator for the treatment, cure, prevention, or slowing of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease, or any other neurodegenerative condition. The mechanistic and preclinical evidence linking NAD+ deficiency to neurodegeneration is largely derived from cell and animal models, and the human evidence is limited to small, early-phase trials that have not demonstrated disease modification. Nothing here is medical advice or a recommendation for human use. Readers should consult qualified healthcare professionals and applicable regulations before making any health decisions.