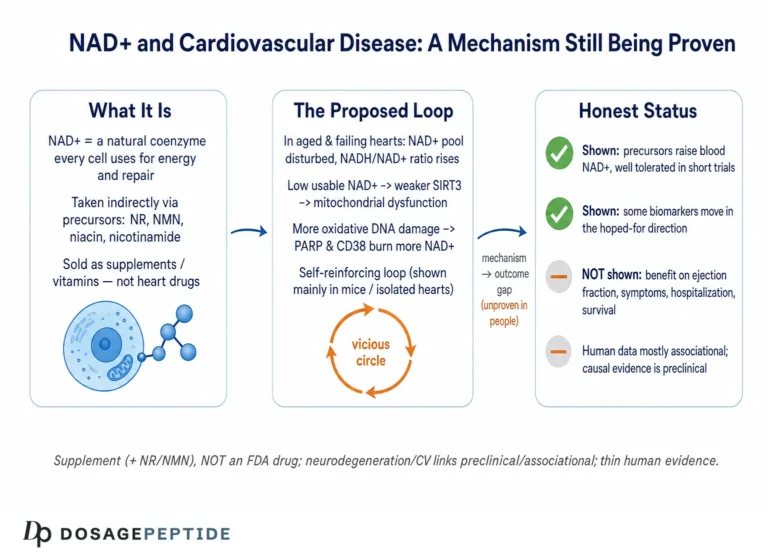
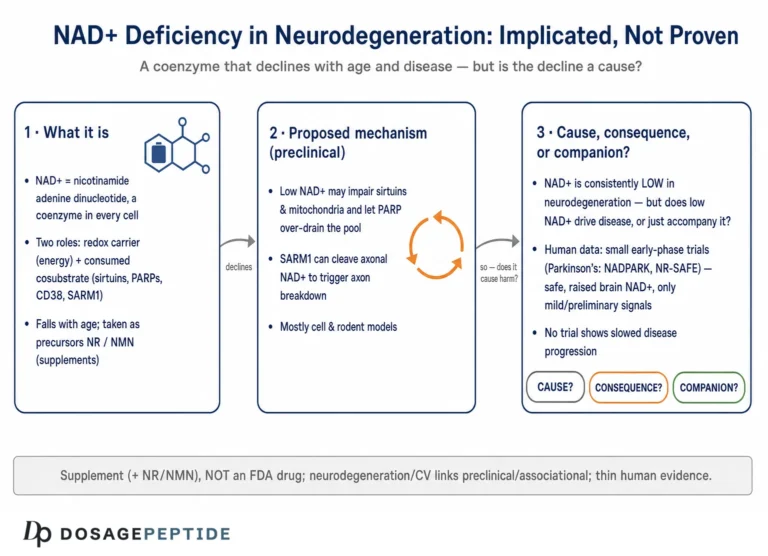
The title asks how NAD+ depletion influences molecular pathways in neurodegenerative disease progression — a phrasing that quietly assumes three things worth separating before we go further: that NAD+ meaningfully falls in the diseased brain, that this fall is a driver rather than a bystander, and that the pathways it touches actually push disease forward rather than merely marking that something has gone wrong. Each of those is a distinct empirical claim, and they are not equally well supported. So rather than treat the premise as settled, this article maps it honestly: where the mechanism is strong and biochemically specific, where it is plausible but correlational, and where the popular narrative has run ahead of the data.
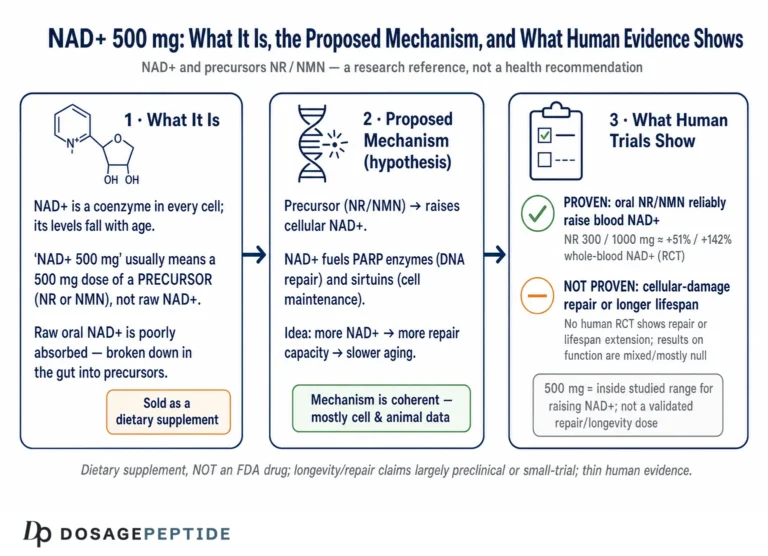
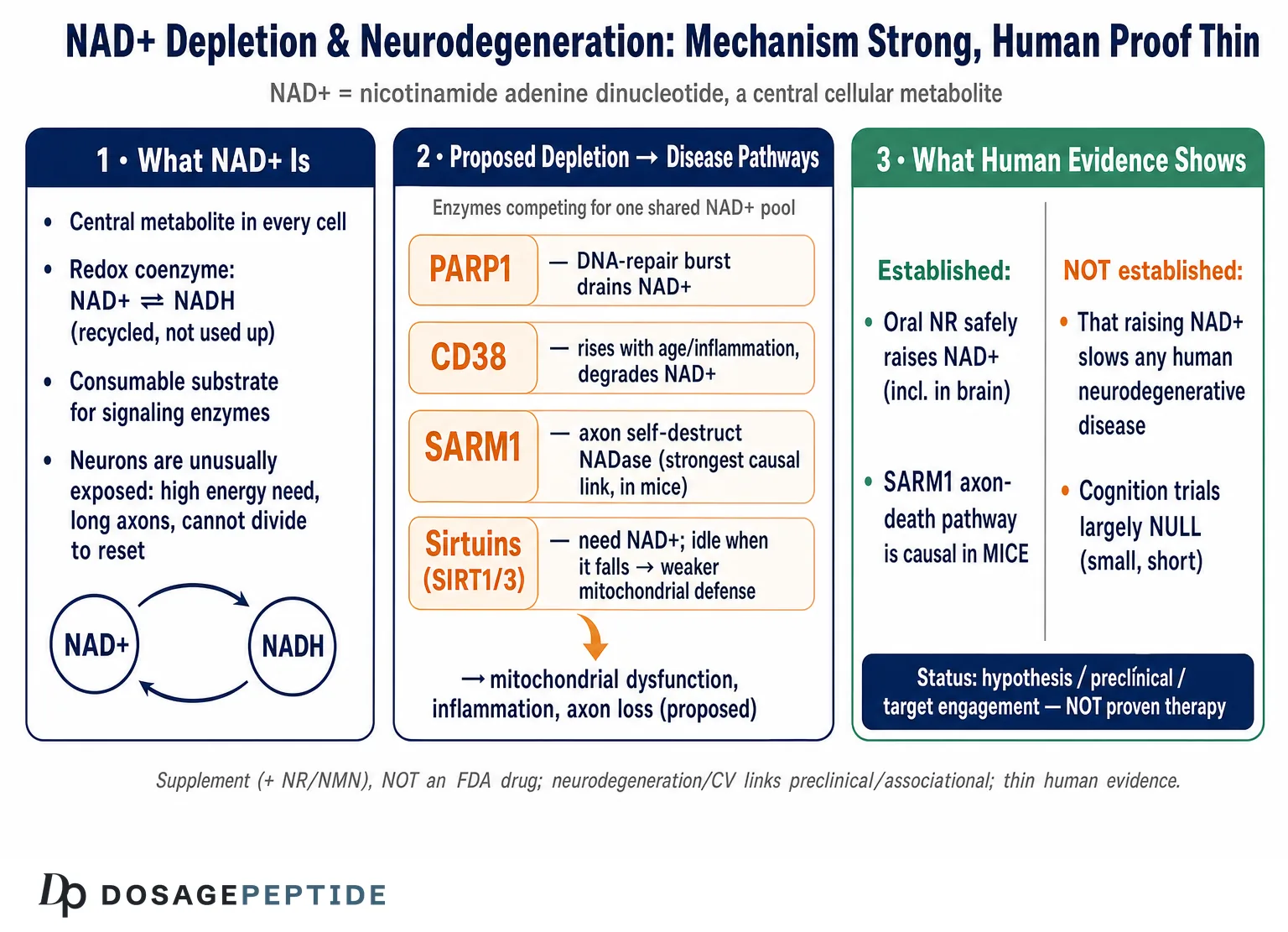
Here is the short version, stated up front so nothing that follows is oversold. NAD+ (nicotinamide adenine dinucleotide) is a genuinely central metabolite, and there is robust molecular biology linking its consumption to axon degeneration, mitochondrial function, DNA repair, and inflammation.12 That much is real cell biology, much of it worked out in exquisite detail. What is not established is that raising NAD+ — by swallowing precursors such as nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN) — slows, halts, or reverses any human neurodegenerative disease. NR and NMN are sold as dietary supplements. They are not FDA-approved drugs for Alzheimer’s, Parkinson’s, ALS, or any other condition. The human trials that exist are small, short, and designed to test safety and target engagement, not to prove clinical benefit.8910 Keeping those two registers apart — the strong mechanistic story and the thin therapeutic evidence — is the single most important habit for reading this field clearly.
This piece is written for researchers and scientifically literate readers who want an accurate map of what NAD+ depletion does at the molecular level, which of those effects plausibly feed disease progression, and how far the human evidence actually reaches. We will build up the biochemistry, then examine each proposed pathway on its own merits, then confront the causation gap and the regulatory reality. Throughout, the guiding rule is restraint: nothing here should be read as a claim that NAD+ or its precursors treat, cure, or prevent neurodegenerative disease.
A word on why the honest framing matters practically, not just ethically. NAD+ occupies an unusual position in the current landscape: it is simultaneously the subject of first-rate molecular biology published in the best journals and the marketing hook for a booming consumer-supplement industry. Those two worlds share vocabulary but not standards of proof, and claims migrate freely from the first to the second, shedding their caveats along the way. A finding that “NAD+ repletion rescued memory in a DNA-repair-deficient mouse” becomes, three commercial steps later, “NAD+ protects your brain from Alzheimer’s.” The mechanistic material in this article is exactly the raw material such claims are built from, so it is worth reading it as a scientist would — asking, at every step, whether a result was obtained in a dish, a mouse, or a person, and whether it measured a biomarker or an actual clinical outcome. Those two questions dissolve most of the overreach on their own.
What NAD+ Is and Why Neurons Are Unusually Exposed
NAD+ is a small dinucleotide that every cell uses in two fundamentally different ways, and the distinction matters for everything that follows. In its first role it is a redox coenzyme: it shuttles electrons between reactions, cycling between oxidized (NAD+) and reduced (NADH) forms, and this cycle is the arithmetic backbone of glycolysis, the citric-acid cycle, and oxidative phosphorylation. In this role NAD+ is not consumed — it is regenerated over and over, so the pool turns without net loss. In its second role NAD+ is a consumable substrate: a family of enzymes cleaves the molecule at its glycosidic bond, using the ADP-ribose moiety for signaling and releasing nicotinamide as a byproduct. This second role is destructive to the pool, and it is the reason NAD+ levels can fall at all.12
Three classes of enzyme dominate that consumption: the sirtuins (NAD+-dependent deacylases, SIRT1–7), the poly(ADP-ribose) polymerases (PARPs, above all PARP1), and the NAD+ glycohydrolases (chiefly CD38, and in axons the injury sensor SARM1). Because these enzymes draw on a shared pool, they are effectively in competition: heavy demand from one — a PARP1 burst after DNA damage, say — can starve the others, so a fall in NAD+ is not just a fuel shortage but a reshuffling of which signaling programs can run.2 This competitive coupling is the conceptual key to the whole depletion hypothesis, and we will return to it repeatedly.
Why are neurons especially exposed? Several features stack up. They are among the most energy-hungry cells in the body and rely heavily on mitochondrial oxidative phosphorylation, so they are sensitive to any wobble in the NAD+/NADH redox ratio. They are post-mitotic and long-lived, meaning they cannot dilute accumulated damage by dividing and must instead repair it in place, which taxes NAD+-consuming repair enzymes over decades. And their architecture is extreme: a single motor neuron can maintain an axon a meter long, and that distal axonal compartment depends on a locally synthesized, rapidly turned-over supply of NAD+ that, as we will see, is uniquely vulnerable to collapse.14 The brain, in short, is a tissue whose cells combine high NAD+ demand, poor tolerance for depletion, and no option to reset by proliferating — which is exactly why NAD+ biology has become a magnet for neurodegeneration researchers. That interest is legitimate; whether it translates into therapy is the open question.
The Depletion Itself: What Actually Falls, and Who Is Doing the Cutting
Before asking what NAD+ depletion does, it is worth being precise about the depletion itself, because loose talk here is the origin of much overreach. The best-characterized fact is that tissue NAD+ declines with age across many organs, and the dominant reason appears to be increased consumption rather than failed synthesis.23 The landmark demonstration came from work showing that the NADase CD38 rises with age and that CD38-knockout mice are largely protected from the age-related NAD+ decline; at 32 months, wild-type mice had roughly half the NAD+ of young animals, while CD38-null mice showed essentially no drop.3 CD38 degrades NAD+ (and its precursor NMN) into ADP-ribose and cyclic ADP-ribose, and its age-linked upregulation — driven substantially by inflammatory and senescence signals — makes it a principal architect of the aging NAD+ deficit.3
Layered onto that baseline age-related decline are disease-specific consumers. PARP1 is activated by DNA strand breaks and, when DNA damage is chronic, can hyperconsume NAD+ in a bid to keep up with repair. SARM1, dormant in healthy axons, becomes a voracious NADase once its metabolic brake is released. Neuroinflammation recruits CD38-expressing microglia and macrophages that further drain the local pool.12 The picture that emerges is not a single leak but several taps, each opened by a different pathological trigger, all draining the same reservoir.
Two honest caveats belong here. First, measuring NAD+ in the living human brain is hard; most human data come from blood, cerebrospinal fluid, or post-mortem tissue, each an imperfect proxy for what a living neuron’s cytosolic or mitochondrial pool is doing in real time. The one clean demonstration that an oral precursor raises cerebral NAD+ in living patients came from a Parkinson’s trial using 31-phosphorus magnetic resonance spectroscopy, and even there the increase was significant but variable across individuals.8 Second, subcellular compartmentation matters enormously: NAD+ in the nucleus, cytosol, and mitochondria is separately regulated, and a whole-tissue average can hide a collapse in one compartment or a surplus in another. When a headline says “brain NAD+ is low in disease,” the rigorous reader should ask which pool, measured how, in whom. For readers tracking how these measurements have been made in human cohorts specifically, the companion discussion of how NAD 500 mg influences cellular damage repair and longevity in human studies catalogs the dosing-and-measurement side of this literature.
Mitochondrial Bioenergetics and the Sirtuin Axis
The most intuitive way NAD+ depletion could drive neurodegeneration is bioenergetic: less NAD+ means a disturbed NAD+/NADH ratio, which slows the electron flux through oxidative phosphorylation and lowers ATP output in cells that can least afford it. This is real, but it is also the crudest version of the story, and on its own it is probably not the whole mechanism, because the redox pool is regenerative and cells defend it vigorously. The more interesting and specific pathway runs through the sirtuins, the NAD+-dependent deacylase enzymes whose activity is gated directly by NAD+ availability.12
Because sirtuins consume NAD+ as an obligatory co-substrate, their activity is a readout of the cell’s metabolic state: when NAD+ is plentiful they are active; when it falls they idle. That coupling matters because several sirtuins sit at control points for the very processes that fail in neurodegeneration. SIRT1 deacetylates and activates PGC-1α, the master regulator of mitochondrial biogenesis and antioxidant defense; when NAD+ drops and SIRT1 slows, PGC-1α signaling weakens, and the cell makes fewer, less capable mitochondria at the moment it needs more.1 SIRT3, the principal mitochondrial sirtuin, deacetylates enzymes of the respiratory chain and antioxidant system; SIRT3 levels are reported to fall in Alzheimer’s cortex, with a corresponding rise in acetylated, dysfunctional mitochondrial proteins, tying NAD+/SIRT3 decline to the mitochondrial pathology seen in the disease.1 SIRT3 activity also depends partly on CD38, closing a loop in which the age-related NADase both lowers NAD+ and, through that, disables a key protective deacylase.3
The mechanistic logic here is genuinely appealing: NAD+ depletion does not merely dim the lights on energy production, it silences a stress-response and quality-control program (sirtuin-driven mitophagy, biogenesis, and antioxidant induction) precisely when a stressed neuron is trying to switch it on. A defect in mitophagy — the selective clearance of damaged mitochondria — is one of the more compelling threads here, because impaired removal of failing mitochondria would let dysfunctional organelles accumulate, spilling reactive oxygen species and further degrading the NAD+ pool; systematic reviews of the in vivo Alzheimer’s literature single out this NAD+–mitophagy connection as one of the better-supported preclinical rationales for the whole approach.13 In animal and cell models, restoring NAD+ with precursors reactivates this axis and improves mitochondrial readouts.7 But two disciplines are required. First, most of this evidence is preclinical — rodent brains, cultured neurons, isolated mitochondria — and rodent NAD+ biology does not map one-to-one onto humans. Second, “improves a mitochondrial biomarker” is not the same as “slows a disease”; the history of neurodegeneration research is littered with agents that fixed a mechanism in a dish and did nothing in a patient. The sirtuin axis is the strongest bioenergetic case for the depletion hypothesis, and it is still, in humans, an unproven one.
SARM1 and Axon Degeneration: the Sharpest Mechanistic Link
If any part of the NAD+–neurodegeneration story deserves the word “mechanism” without hedging, it is the SARM1 axon-degeneration pathway — a piece of biology so clean it has reoriented the whole field. The story begins with an old genetic curiosity: the Wallerian degeneration slow (WldS) mutant mouse, whose severed axons survive far longer than normal because the mutation ectopically supplies the NAD+-synthesizing enzyme NMNAT1 to axons.4 That clue pointed at NAD+ metabolism as the executioner of axon death, and the pieces fell into place over the following years.
In a healthy axon, the labile enzyme NMNAT2 is continuously transported outward and continuously degraded; its job is to convert NMN into NAD+, keeping the local NMN low and NAD+ adequate. When an axon is injured — or when transport fails, or mitochondria falter — NMNAT2 supply collapses. NMN then accumulates and NAD+ falls, and it is this shift in the ratio, not the absolute NAD+ level alone, that trips the sensor.56 The sensor is SARM1. In a discovery that upended textbook assumptions, SARM1’s TIR domain was shown to be not a scaffolding module but an enzyme — an intrinsic NADase that cleaves NAD+ into nicotinamide, ADP-ribose, and cyclic ADP-ribose.4 Structural and biochemical work then showed that a rising NMN/NAD+ ratio binds an allosteric pocket on SARM1’s regulatory domain, releasing the brake and unleashing runaway NAD+ destruction.5 The result is a catastrophic, self-amplifying local NAD+ collapse that dismantles the axon.
What makes this the field’s crown jewel is the causal proof: genetically deleting or inhibiting SARM1 dramatically protects axons across a striking range of insults — traumatic and chemotherapy-induced neuropathy, certain glaucoma and traumatic-brain-injury models, and features of ALS and other neurodegenerative conditions.45 Here, NAD+ depletion is not correlated with degeneration; it is the degeneration program, executed enzymatically. This is why SARM1 inhibitors are in active drug development as a genuinely mechanism-based therapeutic strategy for axonopathies.
It is worth pausing on why this pathway reframes the entire relationship between NAD+ and neurodegeneration. In the classical bioenergetic view, an axon dies passively when it runs out of energy — degeneration as a slow starvation. The SARM1 discovery replaced that picture with an active, tightly regulated program: the axon is not a candle guttering out but a structure carrying a loaded self-destruct switch, held in check by a continuous supply of NMNAT2 and released the moment that supply fails. Degeneration is thus something the axon does, not merely something that happens to it. That distinction is not academic. A passive-starvation model predicts that adding fuel (NAD+ or its precursors) should help; an active-program model predicts that the decisive lever is the switch itself, and that the pharmacology should aim to keep SARM1 off — which is exactly the strategy the drug-development field has adopted. The same logic explains why NMNAT2, the labile enzyme whose loss releases the brake, is itself now viewed as a neuroprotective factor whose stabilization could, in principle, keep the program suppressed.6
But precisely because this pathway is so well understood, it also exposes a paradox that undercuts naive “just take more NAD+” reasoning. The trigger for SARM1 is a high NMN/NAD+ ratio, and NMN is a direct NAD+ precursor. In principle, flooding a compromised axon with NMN could raise the very ratio that activates the destructive enzyme, while the protective strategy is to lower NMN (for example by inhibiting the enzyme NAMPT that makes it, or by inhibiting SARM1 directly) rather than to pour in more precursor.56 The relationship between NAD+ metabolites and axon survival is thus not a simple “more is better” dial; it is a ratio-sensitive control system in which the wrong intervention could plausibly do nothing or worse. This nuance is routinely lost in consumer messaging, and it is a clean illustration of why mechanistic depth argues for humility, not confidence, about supplementation. The parallel question of whether raising NAD+ can protect neurons in an acute setting is examined in the sibling analysis of what scientific evidence supports NAD in preserving neurons during stroke, where the same “mechanism strong, human proof thin” tension recurs.
DNA Repair, PARP1 Hyperactivation, and the Genomic-Stress Loop
A second specific pathway links NAD+ depletion to neurodegeneration through the genome. Neurons accumulate DNA damage over a lifetime, and the enzyme PARP1 is the front-line detector of DNA strand breaks: it binds damaged DNA and synthesizes chains of poly(ADP-ribose) onto target proteins to recruit the repair machinery. The raw material for those chains is NAD+. Under modest damage this is a fine trade, but under chronic or severe genomic stress PARP1 becomes hyperactive and can consume NAD+ faster than the cell can resynthesize it, driving the pool down.12
This creates a vicious loop that is central to the depletion hypothesis. Falling NAD+ starves the sirtuins (including those that support mitochondrial quality control and DNA-repair fidelity), impairs mitochondrial function, and increases oxidative stress — which produces more DNA damage, which activates more PARP1, which consumes more NAD+. In extreme cases, catastrophic PARP1 activation drives a specific programmed cell-death pathway (parthanatos). The most persuasive experimental support comes from a mouse model combining Alzheimer’s pathology with a DNA-repair deficiency: here, supplementing NAD+ with nicotinamide riboside normalized several disease features — reducing neuroinflammation, restraining phosphorylated tau, improving DNA-damage responses, and rescuing learning, memory, and motor function.7 That study is frequently — and fairly — cited as proof of principle that the NAD+–DNA-repair loop is manipulable.
There is a deeper reason the brain is exposed to this particular loop. Because neurons do not divide, they cannot dilute mutations or offload damaged genomes to daughter cells; every strand break must be repaired in place, and the repair itself is metabolically expensive in NAD+ terms. Over a human lifespan of eighty or ninety years, a neuron may run its PARP-dependent repair machinery countless times, and any age-related shortfall in NAD+ resynthesis makes each subsequent repair episode more costly to the pool. This is why the DNA-repair axis is often framed as an accelerant of aging biology rather than a discrete disease mechanism: it converts the ordinary accumulation of genomic wear into a progressive metabolic tax that compounds with time and interacts with the mitochondrial and inflammatory pathways already described.
The honest framing, though, is that this is a rodent proof of principle in a genetically engineered model built specifically to make the DNA-repair axis dominant. It demonstrates that the loop exists and can be broken in mice; it does not demonstrate that the same lever moves human Alzheimer’s, where the damage is multifactorial and the DNA-repair contribution is one voice in a large chorus. The same PARP1–NAD+–sirtuin logic also connects NAD+ to genomic maintenance far beyond the brain, a thread developed in the related discussion of what mechanisms link NAD to DNA repair and cancer prevention. For the neurodegeneration question, the DNA-repair pathway earns its place as a plausible, experimentally supported contributor to progression — while remaining, in humans, a hypothesis rather than a demonstrated therapeutic target.
Neuroinflammation, CD38, and the Senescence Loop
The third major axis connecting NAD+ depletion to disease progression is inflammatory, and it has tightened considerably in recent years. Chronic, low-grade neuroinflammation — “inflammaging” — is a feature of both brain aging and neurodegenerative disease, and it turns out to be tightly wired to NAD+ metabolism through CD38. Activated microglia and infiltrating macrophages upregulate CD38, and CD38 is a major NAD+ consumer; so an inflamed brain is, by that fact alone, an NAD+-depleting environment.23
The loop closes through cellular senescence. Senescent cells accumulate with age and adopt a pro-inflammatory secretory phenotype; that secretome recruits and activates CD38-expressing macrophages, which then drain tissue NAD+.2 Direct experimental work has shown that CD38 expression in immune cells is induced during aging and that these CD38-positive macrophages are a major sink for both NAD+ and its precursor NMN, degrading them in the surrounding tissue.12 The consequences run in both directions: falling NAD+ compromises the sirtuin-dependent restraint of inflammatory signaling (SIRT1, for instance, normally dampens NF-κB-driven transcription), so depletion both results from and amplifies inflammation. In the brain specifically, microglia are the resident immune cells, and their shift from a surveilling to an activated, CD38-high state during neurodegeneration would be expected to lower local NAD+ at exactly the sites — synapses and their supporting glia — where energy demand and repair needs are highest. This positions the inflammatory axis not as a separate story from the mitochondrial and DNA-repair pathways but as a hub that feeds both: an inflamed microenvironment lowers the NAD+ available to sirtuins and repair enzymes throughout the affected tissue. In the specific setting of Alzheimer’s models, NAD+ repletion has been reported to reduce neuroinflammation and cellular senescence, in part by quieting the cGAS–STING innate-immune sensing pathway.11 This gives a coherent molecular narrative in which NAD+ depletion is simultaneously a symptom of the inflamed, senescent brain and a contributor to its further decline.
As with the other axes, the mechanism is real and the therapeutic implication is speculative. It is entirely plausible that lowering CD38 activity would be a more logical intervention than adding precursors — if you are losing NAD+ because an enzyme is destroying it, inhibiting that enzyme addresses the cause, whereas adding substrate may just feed the same consumer. CD38 inhibitors are, accordingly, an area of active preclinical interest. None of this has been shown to alter the course of a human neurodegenerative disease. The value of the inflammatory axis, for now, is explanatory: it helps account for why brain NAD+ falls in disease and aging, and it reframes the deficit as partly a consequence of immune dysregulation rather than a primary metabolic failure.
Disease by Disease: Where Association Is Strong and Causation Is Not
It is tempting to treat “neurodegenerative disease” as one thing, but the strength of the NAD+ evidence varies markedly across conditions, and lumping them together is a common source of overstatement. The table below is a candid ledger of how far the evidence actually reaches for each, kept deliberately conservative.
| Condition | Proposed NAD+-linked mechanism | Strongest evidence level |
|---|---|---|
| Alzheimer’s disease | PARP1/DNA-repair drain, SIRT3 loss & mitochondrial dysfunction, CD38-driven neuroinflammation; possible SARM1-linked synapse loss1711 | Mouse models + small human NR trial with mixed/negative cognitive results710 |
| Parkinson’s disease | Mitochondrial (complex I) dysfunction, impaired mitophagy, low cerebral NAD+; NR raises brain NAD+18 | Two small phase I human trials (target engagement + safety), no proven disease modification89 |
| ALS / peripheral neuropathy | SARM1-executed axon degeneration; NMNAT2 loss; NMN/NAD+ ratio trigger45 | Strong causal genetics in mice; SARM1 inhibitors in development; no approved therapy |
| Huntington’s disease | Sirtuin dysregulation, mitochondrial energetic failure1 | Preclinical and associational only |
| Age-related cognitive decline | Systemic NAD+ decline via CD38; sirtuin idling23 | Association in humans; small NR trials largely null on cognition10 |
Two patterns stand out. First, the mechanistic confidence is highest precisely where the human therapeutic evidence is absent: the SARM1 axon pathway is causally airtight in mice, yet there is no approved NAD+-based therapy for any axonopathy, and the logical intervention there is inhibition of an NADase rather than precursor supplementation. Second, where human trials do exist — Parkinson’s and Alzheimer’s — they are small, short, and were designed to answer narrow questions (is it safe, does it raise NAD+), not to demonstrate that progression slows. The gap between “NAD+ depletion is mechanistically entangled with this disease” and “correcting NAD+ changes the disease’s course in people” is, in every row of that table, unbridged. The Parkinson’s-specific version of this ledger is developed further in the sibling article on the link between NAD deficiency and Parkinson’s progression.
From Mechanism to Human Data: What the Trials Actually Found
Because the mechanistic literature is so seductive, the fairest test of the depletion hypothesis is to look squarely at what happened when precursors were given to actual patients. The human record is small but instructive, and its most honest reading is “target engaged, benefit unproven.”
The NADPARK study — a randomized, double-blind, placebo-controlled phase I trial — gave 30 newly diagnosed, drug-naive Parkinson’s patients 1,000 mg of nicotinamide riboside or placebo for 30 days.8 Its headline achievement was target engagement: using 31-phosphorus magnetic resonance spectroscopy, the investigators showed a significant (if variable) increase in cerebral NAD+ in the NR group — a genuinely important demonstration that an oral precursor can reach the human brain and move the metabolite it is supposed to move. Patients whose brain NAD+ rose showed altered cerebral metabolism on FDG-PET and a mild clinical improvement.8 That is a legitimately encouraging phase I signal. It is also, by design, not evidence of disease modification: 30 patients, 30 days, no long-term or functional endpoints, and an explicitly exploratory clinical readout. The follow-up NR-SAFE trial then established that much higher doses — up to 3,000 mg daily for 30 days — are safe and robustly augment the NAD+ metabolome without depleting methyl donors, clearing the way for larger dose-optimization and efficacy studies that had not, at the time of writing, reported disease-modifying results.9
The Alzheimer’s side is more sobering. A crossover, double-blind, randomized placebo-controlled trial tested 1 g/day of NR for eight weeks in older adults with subjective cognitive decline and mild cognitive impairment, using a validated cognitive battery (the Repeatable Battery for the Assessment of Neuropsychological Status, RBANS) as the primary outcome and a panel of plasma Alzheimer’s biomarkers — phosphorylated-tau 217 (pTau217), glial fibrillary acidic protein (GFAP), and neurofilament light chain (NfL) — as secondary measures.10 The result was null on the primary endpoint: NR did not improve cognition on either the conventional battery or digital assessments. The one notable secondary signal was a statistically significant relative reduction in plasma pTau217 (approximately a 7% fall on NR versus an 18% rise on placebo, p = 0.02), while GFAP and NfL were unchanged.10 A single biomarker of tau pathology moving favorably while cognition does not is a hypothesis-generating result, not a demonstration of clinical benefit; the authors’ own conclusion was appropriately modest — a larger, longer trial would be needed to determine whether NR meaningfully alters the Alzheimer’s trajectory — which is precisely the language of a hypothesis not yet confirmed.10 The table below summarizes the human record without embellishment.
| Trial | Design | What it showed | What it did NOT show |
|---|---|---|---|
| NADPARK (Parkinson’s)8 | RCT, phase I; 30 patients; NR 1,000 mg/day × 30 days | Oral NR raised cerebral NAD+ (variable); mild exploratory clinical/metabolic signal | No disease modification; tiny, brief, exploratory endpoints |
| NR-SAFE (Parkinson’s)9 | RCT, phase I safety; 20 patients; up to NR 3,000 mg/day × 30 days | High-dose NR safe; strong NAD+ metabolome augmentation; no methyl-donor depletion | Not powered for efficacy; a safety/dose study only |
| NR in SCD/MCI (Alzheimer’s)10 | RCT crossover; NR 1 g/day × 8 weeks; RBANS primary | Well tolerated; significant relative drop in plasma pTau217 vs placebo | Null on cognition; GFAP/NfL unchanged; no proven clinical benefit; needs larger trial |
Put together, the human evidence supports exactly two firm conclusions and no more. First, oral NR reliably and safely raises the NAD+ metabolome, including in the brain — the pharmacology works. Second, no trial to date has demonstrated that this translates into slowed progression of any neurodegenerative disease. Anyone who tells you the trials “show NAD+ fights Alzheimer’s and Parkinson’s” is reading press releases, not endpoints; the distance between a significant change in a metabolite and a meaningful change in a patient’s disease trajectory is precisely the distance these small studies were never built to cross. A related, non-neurological approach to age-associated cognitive decline — growth-hormone secretagogue signaling — is examined for the same evidentiary caution in the discussion of whether sermorelin supports cognitive function in age-related neurodegeneration, and the pattern is the same: mechanistic plausibility outrunning human proof.
Precursors, Compartmentation, and Why “Raising NAD+” Is Not Simple
A recurring assumption in consumer discourse is that because NAD+ falls, and because precursors raise it, correcting the deficit must help. Several layers of biology complicate that syllogism, and taking them seriously is part of honest scientific communication.
First is the question of which pool rises. NAD+ is salvaged and synthesized through distinct routes — the salvage pathway (nicotinamide → NMN via NAMPT → NAD+ via NMNATs), the NR route (NR → NMN via NRKs), and de novo synthesis from tryptophan — and these feed nuclear, cytosolic, and mitochondrial pools that are separately regulated.2 An oral precursor that raises whole-blood or even whole-brain NAD+ has not necessarily corrected the specific subcellular deficit that matters in a given neuron. Bulk repletion and targeted repletion are different achievements, and current supplements deliver the former.
Second is the SARM1 paradox already discussed: in the axon-degeneration pathway, it is a high NMN/NAD+ ratio that is pathogenic, and NMN is a precursor.56 Whether a given supplementation strategy raises NAD+ enough to lower that ratio, leaves it unchanged, or transiently worsens it depends on kinetics that are not well characterized in diseased human tissue. This is a concrete example of how the same molecule can be protective or provocative depending on the pathway and the compartment.
Third is the consumer-versus-substrate problem. If NAD+ is low largely because CD38 or PARP1 is destroying it, adding substrate may simply feed the destroyer while leaving the underlying dysregulation intact. Mechanistically, the more targeted strategies — inhibiting CD38, inhibiting SARM1, or restraining PARP1 hyperactivation — address the cause of depletion rather than trying to out-supply it.34 None of these are available as approved neurodegeneration therapies, and precursor supplementation persists in the market largely because it is safe, cheap, and available, not because it has out-competed these alternatives on evidence. For readers who encounter NAD+ handling and dosing frameworks in a research context, the site’s central dosages index and peptide and metabolite glossary catalog these compounds for educational reference rather than as guidance for human use.
The upshot is that “raising NAD+” is a genuinely blunt instrument aimed at a finely compartmentalized, ratio-sensitive, multi-consumer system. That does not make it useless as a research probe. It does mean that the intuitive leap from “NAD+ is low in the disease” to “take NAD+ precursors to treat the disease” skips over most of the biology that determines whether such an intervention could work at all.
Limitations and the Causation Gap
Pulling the threads together, the limitations that bear on the title’s question are substantial and they interact, so it is worth stating them plainly rather than burying them.
Association is not causation, and the direction is often unclear. In human disease, low NAD+ frequently travels with mitochondrial dysfunction, inflammation, and DNA damage — but these are the same processes that consume NAD+. A neuron that is sick for other reasons will show low NAD+ as a consequence, not necessarily a cause. Distinguishing NAD+ depletion as a driver of progression from NAD+ depletion as a marker of a cell already in trouble is genuinely hard, and much of the human evidence cannot make that distinction at all.
The strongest causal biology does not endorse supplementation. The one place where NAD+ metabolism is proven to cause neurodegeneration — the SARM1 axon-death pathway — points toward enzyme inhibition and toward lowering a metabolite ratio, not toward flooding the system with precursors.45 The mechanism most people cite as proof that NAD+ “matters” is, on close reading, an argument for a different therapeutic strategy than the one being sold.
Human trials are small, short, and target-engagement-focused. The best human data show that precursors raise NAD+ safely.89 The trials designed to test cognition have been largely null.10 No adequately powered, long-duration, disease-modification trial has reported a positive result in any neurodegenerative condition. That is not evidence of failure — it is the absence of the evidence that would be needed to make a therapeutic claim.
Rodent-to-human translation is unreliable here specifically. Baseline NAD+ turnover, precursor pharmacokinetics, lifespan, and disease biology all differ between mice and people, and the field’s most cited positive results come from genetically engineered mouse models built to amplify one pathway.7 These are proofs of concept, not previews of clinical outcome.
Measurement and compartmentation confound interpretation. Most human NAD+ data are peripheral proxies, subcellular pools are separately regulated, and even the one clean brain measurement showed marked person-to-person variability in response.8 Confident statements about “the brain’s NAD+” usually paper over these limits.
The causation gap, then, is not a footnote — it is the center of the honest answer to the title. NAD+ depletion is deeply entangled with the molecular pathways of neurodegeneration, and in the axon-degeneration case it is a proven executioner. But “entangled with” and “proven executioner in mice” are not the same as “correctable driver of human disease progression by supplementation,” and the responsible position is to hold those apart.
Regulatory and Safety Status
Because the science is so often discussed alongside products for sale, the regulatory reality deserves explicit statement. NAD+ itself and its precursors nicotinamide riboside and nicotinamide mononucleotide are marketed in the United States as dietary supplements, not as drugs. They are not approved by the FDA, the EMA, or any comparable regulator for the treatment, prevention, or cure of Alzheimer’s disease, Parkinson’s disease, ALS, Huntington’s disease, age-related cognitive decline, or any other neurodegenerative condition. There is no approved neurodegeneration indication for any NAD+-based product, and dietary-supplement status specifically means a product has not been evaluated by the FDA for efficacy against any disease.
The precursors have been studied enough to say something reassuring about short-term safety in the populations tested: NR was well tolerated in Parkinson’s patients even at 3,000 mg daily over a month, with no depletion of methyl donors and no serious adverse events.9 That is a real and useful finding — but short-term tolerability in a small trial is not the same as long-term safety across the years-to-decades horizon over which a neurodegenerative disease unfolds, nor across the frail, poly-medicated, elderly populations most affected. It is also worth noting that the regulatory status of NMN in the United States has been contested, with the FDA taking the position that NMN is excluded from the dietary-supplement definition because it has been investigated as a drug — a live regulatory dispute that further underscores the unsettled ground these products occupy.
The synthesis is straightforward: NAD+ precursors sit in the dietary-supplement category, backed by strong mechanistic biology, encouraging target-engagement data, and no proof of clinical benefit for any neurodegenerative disease. Any legitimate investigation of NAD+ modulation as a neurodegeneration therapy belongs in properly designed, adequately powered clinical trials under regulatory oversight — not in off-label inference from mechanism or from supplement marketing.
Frequently Asked Questions
Does NAD+ depletion cause neurodegenerative disease?
In one specific and well-proven case — axon degeneration executed by the enzyme SARM1 — NAD+ destruction is genuinely causal, and blocking that pathway protects axons in animal models.45 More broadly, however, low NAD+ in the diseased human brain is at least partly a consequence of the mitochondrial dysfunction, inflammation, and DNA damage that also consume it, so it is often a marker of a cell in trouble as much as a driver.12 Distinguishing driver from marker in humans remains an open problem, and it is why no confident causal claim can be made across neurodegenerative disease in general.
Will taking NR or NMN supplements slow Alzheimer’s or Parkinson’s?
There is no clinical evidence that it does. The human trials completed so far were small and short and were designed to test safety and whether the supplements raise NAD+ — which they do.89 The one cognition-focused Alzheimer’s trial was largely null on its primary endpoints.10 No adequately powered, long-duration disease-modification trial has reported a positive result. NR and NMN are dietary supplements, not approved treatments for any neurodegenerative disease.
What is the single strongest piece of NAD+–neurodegeneration biology?
The SARM1 axon-degeneration pathway. SARM1’s TIR domain is an NADase that, when unleashed by a rising NMN/NAD+ ratio, destroys axonal NAD+ and triggers self-amplifying axon breakdown; deleting or inhibiting SARM1 protects axons across many injury and disease models.45 This is real, causal, mechanism-based biology — and notably, it argues for inhibiting an enzyme rather than for taking NAD+ precursors.
If NAD+ is low, why not just supplement more of it?
Because the system is more subtle than a fuel gauge. NAD+ exists in separately regulated nuclear, cytosolic, and mitochondrial pools, so raising bulk NAD+ may not fix the specific deficit that matters.2 In the axon pathway, the harmful trigger is a metabolite ratio that a precursor could, in principle, push the wrong way.5 And if an enzyme like CD38 or PARP1 is destroying NAD+, adding substrate may just feed the consumer.3 These are among the reasons the intuitive “more is better” logic is not reliable.
Which enzymes actually deplete NAD+ in the aging and diseased brain?
Three families do most of the consuming: PARPs (chiefly PARP1, activated by DNA damage), the sirtuins (which use NAD+ but are generally protective), and the NADases CD38 and, in injured axons, SARM1.2 CD38 rises with age and inflammation and is a principal driver of the age-related NAD+ decline; CD38-knockout mice are largely protected from that decline.3
Has anyone shown that a precursor raises NAD+ in the living human brain?
Yes. The NADPARK trial used 31-phosphorus magnetic resonance spectroscopy to show that oral nicotinamide riboside significantly increased cerebral NAD+ in Parkinson’s patients — though the response varied substantially between individuals.8 This demonstrates target engagement (the drug reaches the brain and moves the metabolite); it does not demonstrate clinical benefit.
Are NAD+ precursors safe?
In the short-term trials done so far they have been well tolerated, with nicotinamide riboside safe in Parkinson’s patients even at 3,000 mg daily for a month and no depletion of methyl donors.9 That said, short-term tolerability in small trials is not the same as long-term safety over the years a neurodegenerative disease lasts, nor in the frail elderly populations most affected. As dietary supplements, product purity and labeling also vary by source.
Is NAD+ an FDA-approved treatment for any neurological disease?
No. NAD+ and its precursors NR and NMN are sold as dietary supplements and are not approved by the FDA, EMA, or comparable regulators for treating, preventing, or curing any neurodegenerative disease. Dietary-supplement status means the products have not been evaluated for efficacy against disease, and the regulatory status of NMN in particular has been the subject of an ongoing dispute in the United States.
References
- Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019;30(4):630-655. PMID: 31577933. https://pubmed.ncbi.nlm.nih.gov/31577933/
- Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22(2):119-141. PMID: 33353981. https://pubmed.ncbi.nlm.nih.gov/33353981/
- Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23(6):1127-1139. PMID: 27304511. https://pubmed.ncbi.nlm.nih.gov/27304511/
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron. 2017;93(6):1334-1343.e5. PMID: 28334607. https://pubmed.ncbi.nlm.nih.gov/28334607/
- Figley MD, Gu W, Nanson JD, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron. 2021;109(7):1118-1136.e11. PMID: 33657413. https://pubmed.ncbi.nlm.nih.gov/33657413/
- Coleman MP, Höke A. An NAD+/NMN balancing act by SARM1 and NMNAT2 controls axonal degeneration. Neuron. 2021;109(7):1067-1069. PMID: 33831359. https://pubmed.ncbi.nlm.nih.gov/33831359/
- Hou Y, Lautrup S, Cordonnier S, et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci USA. 2018;115(8):E1876-E1885. PMID: 29432159. https://pubmed.ncbi.nlm.nih.gov/29432159/
- Brakedal B, Dölle C, Riemer F, et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022;34(3):396-407.e6. PMID: 35235774. https://pubmed.ncbi.nlm.nih.gov/35235774/
- Bråthen G, Brakedal B, Drågen K, et al. NR-SAFE: a randomized, double-blind safety trial of high dose nicotinamide riboside in Parkinson’s disease. Nat Commun. 2023;14:7793. PMID: 38016957. https://pubmed.ncbi.nlm.nih.gov/38016957/
- Wu J, Singh K, Shing V, et al. Cognitive and Alzheimer’s disease biomarker effects of oral nicotinamide riboside (NR) supplementation in older adults with subjective cognitive decline and mild cognitive impairment. Alzheimers Dement (N Y). 2025;11(1):e70023. PMID: 39817194. https://pubmed.ncbi.nlm.nih.gov/39817194/
- Hou Y, Wei Y, Lautrup S, et al. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci USA. 2021;118(37):e2011226118. PMID: 34497121. https://pubmed.ncbi.nlm.nih.gov/34497121/
- Chini CCS, Peclat TR, Warner GM, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat Metab. 2020;2(11):1284-1304. PMID: 33199925. https://pubmed.ncbi.nlm.nih.gov/33199925/
- Yao Z, Yang W, Gao Z, et al. NAD+ in Alzheimer’s Disease: Molecular Mechanisms and Systematic Therapeutic Evidence Obtained in vivo. Front Cell Dev Biol. 2021;9:668491. PMCID: PMC8369418. https://pmc.ncbi.nlm.nih.gov/articles/PMC8369418/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. NAD+ and its precursors nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are marketed as dietary supplements and are not approved by the FDA, EMA, or any comparable regulator for the treatment, cure, or prevention of Alzheimer’s disease, Parkinson’s disease, ALS, Huntington’s disease, age-related cognitive decline, or any other neurodegenerative condition. The molecular pathways described here are supported largely by preclinical and associational evidence; no human trial has demonstrated that raising NAD+ slows the progression of any neurodegenerative disease. Nothing here is medical advice or a recommendation for human use. Readers should consult qualified professionals and applicable regulations before making any decisions.