The question in the title carries an assumption worth examining before we accept it. To ask how retatrutide research “informs treatment” of atherogenic dyslipidemia implies that retatrutide either is, or is on a clear path to becoming, a therapy for that lipid disorder. That framing runs ahead of the evidence. Retatrutide is an investigational, single-molecule triple agonist of the glucose-dependent insulinotropic polypeptide (GIP), glucagon-like peptide-1 (GLP-1), and glucagon receptors.1 As of mid-2026 it is not approved by the U.S. Food and Drug Administration, the European Medicines Agency, or any comparable regulator for any indication — not for obesity, not for type 2 diabetes, and certainly not for the correction of an atherogenic lipid profile. There has never been a clinical trial in which lowering atherogenic lipoproteins was the primary purpose of giving retatrutide, and there is no completed cardiovascular outcomes trial telling us whether the lipid changes it produces translate into fewer heart attacks or strokes.
So rather than affirm the premise, this article treats it as an open and genuinely interesting research question: does the body of retatrutide data — drawn almost entirely from phase 2 obesity, diabetes, and liver-fat trials, with lipids measured as secondary or exploratory endpoints — tell us anything useful about the biology of atherogenic dyslipidemia, and about whether triple incretin-glucagon agonism might one day be relevant to it? The honest answer, which we will build toward carefully, is that the research is informative in a mechanistic and hypothesis-generating sense, and strikingly so on the specific lipid fractions that define the atherogenic phenotype, but that it stops well short of establishing retatrutide as a treatment for dyslipidemia.2
Atherogenic dyslipidemia — the triad of elevated triglycerides, reduced high-density lipoprotein (HDL) cholesterol, and a preponderance of small, dense low-density lipoprotein (LDL) particles — is not primarily an LDL-cholesterol problem, and that distinction turns out to matter a great deal when interpreting what retatrutide does.2 The two pivotal phase 2 programs, in obesity3 and in type 2 diabetes,4 both reported improvements across exactly the parameters that characterize this triad. Whether that makes retatrutide a plausible future dyslipidemia agent, a useful research probe into triple-agonist lipid biology, or simply another weight-loss drug whose lipid benefits ride on the weight it removes, is the thread we will follow. Throughout, the guiding discipline is restraint: nothing here should be read as suggesting retatrutide treats, cures, or prevents dyslipidemia or cardiovascular disease.
What Atherogenic Dyslipidemia Actually Is
To judge whether retatrutide research is relevant, we first need to be precise about the target, because atherogenic dyslipidemia is frequently and unhelpfully collapsed into “high cholesterol.” It is a distinct pattern, and its distinctiveness is the whole point.
The syndrome is defined by three linked abnormalities: raised fasting triglycerides, low HDL-cholesterol, and a shift in LDL composition toward small, dense particles.2 Crucially, in classic atherogenic dyslipidemia the calculated LDL-cholesterol concentration can be normal or only mildly elevated, which is why relying on LDL-C alone underestimates risk in these patients. The particle that carries the danger is not defined by its cholesterol content but by its number and quality: small, dense LDL penetrates the arterial wall more readily, is retained longer in the subendothelial space, and is more susceptible to oxidation than larger, buoyant LDL.2 Because every atherogenic lipoprotein particle — LDL, intermediate-density lipoprotein, very-low-density lipoprotein (VLDL) remnants, and lipoprotein(a) — carries a single molecule of apolipoprotein B (apoB), the apoB concentration is a direct count of atherogenic particles and a better index of this phenotype than LDL-C.
The engine driving the whole pattern is insulin resistance and its effect on the liver.5 When hepatic insulin signaling is impaired, the liver overproduces large, triglyceride-rich VLDL. That surplus of triglyceride-rich lipoproteins sets off a cascade mediated by cholesteryl ester transfer protein: triglycerides are swapped into LDL and HDL particles in exchange for cholesteryl esters. The triglyceride-enriched LDL and HDL are then acted on by hepatic lipase, which strips away triglyceride and leaves behind small, dense LDL and small HDL particles that are cleared rapidly — hence the low HDL-cholesterol.5 In other words, the elevated triglycerides, the low HDL, and the small dense LDL are not three separate problems; they are three downstream consequences of the same upstream event, hepatic VLDL overproduction driven by insulin resistance. Apolipoprotein C-III, which inhibits lipoprotein lipase and the clearance of triglyceride-rich remnants, sits near the center of this machinery and is itself an emerging marker and mediator of residual cardiovascular risk.
This pathophysiology explains why atherogenic dyslipidemia is the characteristic lipid disturbance of obesity, metabolic syndrome, and type 2 diabetes, and why it contributes to the residual cardiovascular risk that persists even when LDL-cholesterol has been lowered with statins.2 It also frames the key question for any metabolic drug: does the agent act on the upstream driver — hepatic triglyceride and VLDL production, insulin resistance, ectopic liver fat — or merely nudge the downstream numbers? A therapy that genuinely reduced hepatic VLDL output would be expected to lower triglycerides, lower apoB and non-HDL-cholesterol, shift LDL toward larger particles, and modestly raise HDL. Keep that predicted signature in mind, because it is almost exactly what the retatrutide trials reported. Readers who want the receptor-level version of this story can also consult our companion discussion of how GLP-1 pathways regulate lipid metabolism in atherogenic dyslipidemia.
What Retatrutide Is and Why Its Triple Mechanism Matters for Lipids
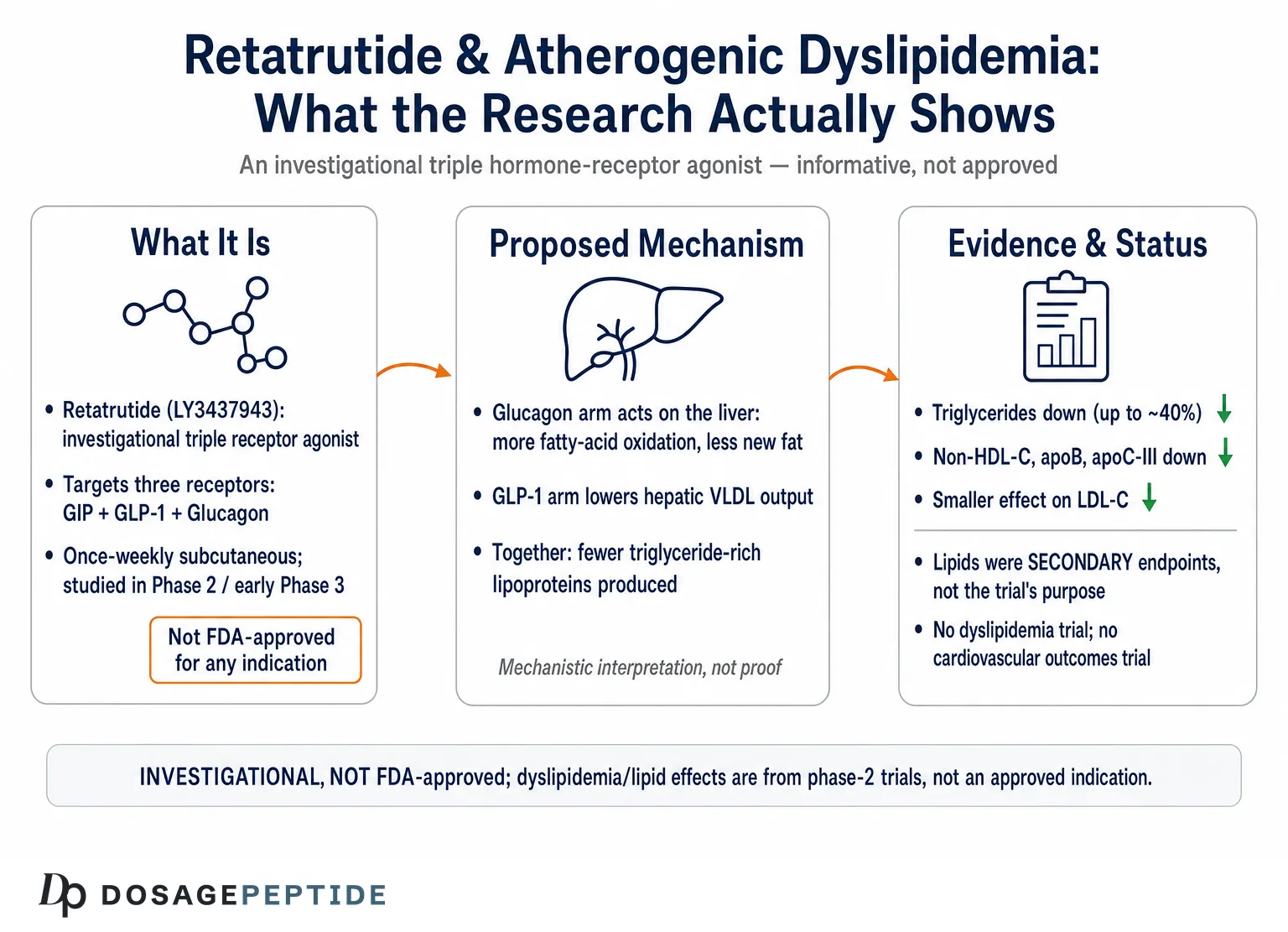
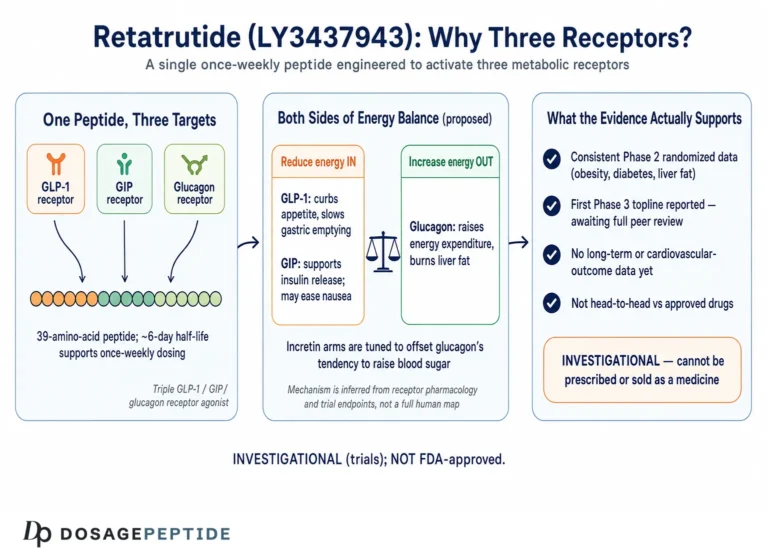
Retatrutide (development code LY3437943) is a synthetic 39-amino-acid peptide built on a glucagon/GIP/GLP-1 sequence scaffold and conjugated to a fatty di-acid moiety that binds albumin and extends its half-life to permit once-weekly subcutaneous administration.1 What makes it pharmacologically distinctive is not that it engages incretin biology — several approved drugs already do — but that it engages three receptors at once, and does so with a deliberately unbalanced potency profile. In cell-based assays, retatrutide is roughly 8.9 times as potent as native GIP at the human GIP receptor, while being less potent than the endogenous ligands at the glucagon and GLP-1 receptors (approximately 0.3 and 0.4 times as active, respectively).1 That tuning matters: it delivers strong GIP-receptor engagement and meaningful, but restrained, glucagon-receptor activity, the latter calibrated to harness glucagon’s metabolic effects on the liver without provoking the hyperglycemia that unopposed glucagon would cause.
Each arm contributes something different to the metabolic and lipid picture. The GLP-1 component suppresses appetite, slows gastric emptying, and improves glucose-dependent insulin secretion — the familiar incretin effects that drive much of the weight loss and glycemic improvement. The GIP component, which retatrutide amplifies, complements GLP-1 on insulin secretion and appears to improve adipose-tissue handling of nutrients and insulin sensitivity. The glucagon component is the genuinely novel ingredient for lipid biology, because glucagon acts directly on the hepatocyte to increase energy expenditure, promote fatty-acid oxidation, and suppress hepatic lipid synthesis. This is the mechanistic reason retatrutide draws attention beyond weight loss, and it is developed further in our overview of retatrutide as the triple-receptor agonist reshaping weight-loss therapies.
It helps to place retatrutide in the lineage of incretin-based drugs. Single GLP-1 receptor agonists (semaglutide, liraglutide) act on one receptor; the dual GIP/GLP-1 agonist tirzepatide adds the GIP axis, which readers can explore in our piece on how tirzepatide influences incretin pathways. Retatrutide adds a third, hepatically-directed receptor. From a lipid standpoint, that third receptor is what changes the story, because it introduces a direct action on the very organ — the liver — whose VLDL overproduction sits at the root of atherogenic dyslipidemia. For a broader view of where this molecule sits historically, see how retatrutide fits into the evolution of peptide therapeutics.
Two cautions are worth stating up front. First, potency at a receptor in a cell line is not the same as the integrated effect in a human being; the clinical lipid results are the real evidence, and mechanism is offered here as interpretation, not proof. Second, and more importantly for the honesty of this article, none of this mechanistic elegance changes retatrutide’s regulatory status: a compelling mechanism for lowering atherogenic lipoproteins is a reason to study a drug, not a reason to claim it works.
The Glucagon Arm: Why a Third Receptor Changes the Lipid Story
Because the glucagon-receptor component is what most differentiates retatrutide from the GLP-1 and GIP/GLP-1 drugs that preceded it, and because its target organ is the liver, it deserves a dedicated look. The lay intuition that glucagon simply “raises blood sugar” is true but radically incomplete, and the incomplete version obscures exactly the part that is relevant to dyslipidemia.
In the hepatocyte, glucagon-receptor activation does more than mobilize glucose. It promotes the mitochondrial oxidation of fatty acids (beta-oxidation) and suppresses de novo lipogenesis, the pathway by which the liver manufactures new fat from carbohydrate substrate.6 The net consequence is a reduction in the hepatic triglyceride pool available for packaging into VLDL, and therefore a reduction in VLDL-triglyceride secretion. Controlled mouse work makes this causal rather than merely associative: glucagon-receptor agonism lowers plasma and liver triglycerides, whereas glucagon-receptor antagonism has the opposite effect, raising them — a clean bidirectional demonstration that the glucagon axis is a genuine regulator of triglyceride metabolism.6 This is precisely the upstream lever that atherogenic dyslipidemia calls for: an intervention that reduces hepatic triglyceride-rich lipoprotein output rather than merely rearranging the downstream particles.
The GLP-1 arm reinforces the same direction of effect through partly independent routes. In insulin-resistant models, GLP-1 receptor agonism reduces hepatic VLDL-apoB overproduction and dampens de novo lipogenesis, contributing to lower circulating triglyceride-rich lipoproteins.7 So in retatrutide the GLP-1 and glucagon components are, for once, pulling the hepatic-lipid lever in the same direction, one largely by reducing the substrate supply and improving insulin sensitivity, the other by directly shifting the hepatocyte toward fat oxidation and away from fat synthesis.
The clearest human fingerprint of this hepatic action comes from the liver-fat data. In a phase 2a trial in adults with metabolic dysfunction-associated steatotic liver disease (MASLD), retatrutide produced dose-dependent relative reductions in liver fat content of approximately 42.9%, 57.0%, 81.4%, and 82.4% at the 1, 4, 8, and 12 mg doses at 24 weeks, versus essentially no change on placebo.8 Reductions of that magnitude — with the majority of participants at higher doses reaching normal liver-fat thresholds — are difficult to attribute to appetite suppression alone and are consistent with a direct hepatic effect on lipid handling. In the same trial, fasting triglycerides fell significantly at doses of 4 mg or greater.8 A liver that is being actively emptied of fat and steered away from lipogenesis is a liver that should export fewer triglyceride-rich lipoproteins, and the circulating lipid data, discussed next, are consistent with that expectation.
It is also worth noting the delicate balancing act the molecule performs. Glucagon-receptor agonism, taken alone, would tend to raise blood glucose — the classic counter-regulatory action of the hormone — which is why a glucagon agonist could not be a metabolic drug on its own. Retatrutide resolves this by pairing restrained, sub-native glucagon-receptor potency with strong GIP and GLP-1 activity that drive glucose-dependent insulin secretion; the incretin arms buffer the glycemic effect of the glucagon arm while the glucagon arm contributes the hepatic energy-expenditure and lipid-oxidation benefits.1 The consequence, seen consistently in the diabetes trials, is that glycemia improves even as the glucagon component does its hepatic work. This is the pharmacological reason a glucagon action that would be harmful in isolation becomes usable, and potentially beneficial for hepatic lipid handling, in the context of a balanced triple agonist.
None of this is a claim that retatrutide corrects dyslipidemia through glucagon action; it is a mechanistic account of why the lipid changes observed in the trials are biologically coherent. Coherence increases plausibility. It does not substitute for the outcome data that would be needed to call retatrutide a lipid therapy.
What the Phase 2 Evidence Actually Shows on Lipids
The strongest human evidence relevant to this question comes from the two large phase 2 trials, in obesity and in type 2 diabetes, where standard lipid parameters were measured as secondary endpoints. Both were randomized, double-blind, and placebo-controlled, and both reported lipid improvements that track the atherogenic-dyslipidemia signature.
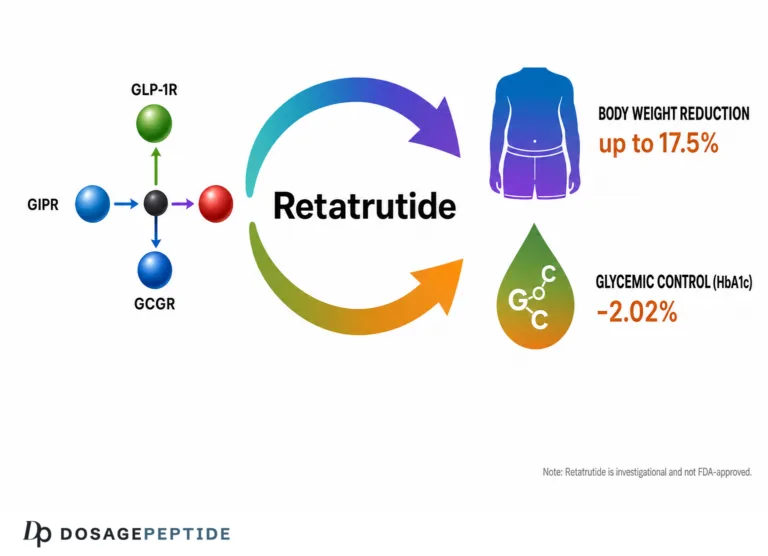
In the obesity phase 2 trial — 338 adults with obesity, or overweight with a weight-related complication, randomized across doses up to 12 mg — retatrutide produced least-squares mean weight reductions of up to 17.5% at 24 weeks and up to 24.2% at 48 weeks, alongside improvements in blood pressure, glycemia, and lipids.3 The type 2 diabetes phase 2 trial enrolled 281 adults (mean HbA1c 8.3%, mean BMI 35.0), compared retatrutide against placebo and against the GLP-1 agonist dulaglutide, and reported dose-dependent HbA1c reductions up to roughly 2.0% and weight reductions up to about 16.9% at higher doses, again accompanied by favorable movements in triglycerides and other cardiometabolic measures.4 The consistency across two different populations — people with obesity and people with type 2 diabetes, the two groups in whom atherogenic dyslipidemia is most prevalent — strengthens the internal validity of the lipid signal.
The table below summarizes what the phase 2 program actually demonstrated on the parameters that matter for atherogenic dyslipidemia. Figures are drawn from the trials and their prespecified and post-hoc lipid analyses; where a single most-informative value exists it is given, and directions are noted where percentages were not uniformly reported.
| Parameter | Direction with retatrutide | Representative magnitude (higher doses) | Relevance to atherogenic dyslipidemia |
|---|---|---|---|
| Triglycerides | Decrease | Up to ~40–41% reduction34 | Core component of the triad; reflects VLDL/remnant load |
| Non-HDL cholesterol | Decrease | Up to ~26.9% reduction9 | Captures all apoB-containing atherogenic particles |
| Apolipoprotein B | Decrease | Up to ~24.2% reduction9 | Direct count of atherogenic particles |
| Apolipoprotein C-III | Decrease | Up to ~38.0% reduction9 | Regulator of remnant clearance; residual-risk marker |
| LDL-cholesterol | Modest / variable decrease | Smaller and less consistent than TG/apoB effects3 | Least representative of this phenotype |
| HDL-cholesterol | Roughly neutral to slight change; HDL particle size increases9 | — | Low HDL is part of the triad; particle quality may matter more than concentration |
| Liver fat (MASLD cohort) | Large decrease | Up to ~82% relative reduction at 24 wk8 | Upstream driver of hepatic VLDL overproduction |
Two features of this pattern deserve emphasis because they are exactly what distinguishes an atherogenic-dyslipidemia effect from a generic “cholesterol-lowering” one. First, the largest effects fall on the triglyceride-rich and apoB-containing fractions — triglycerides, non-HDL-cholesterol, apoB, apoC-III — rather than on LDL-cholesterol, whose movement is comparatively modest and less consistent.3 That is the fingerprint of a drug acting on hepatic triglyceride-rich lipoprotein metabolism, not on the LDL-receptor pathway that statins exploit. Second, the effects are dose-dependent, which is a hallmark of a real pharmacologic action rather than noise.
A necessary caution: these were secondary and exploratory endpoints in trials designed and powered for weight and glycemic outcomes, over 24 to 48 weeks, in selected phase 2 populations. They establish that retatrutide reliably moves atherogenic lipid markers in the favorable direction. They do not establish that it should be used to do so, nor that moving those markers with this drug reduces clinical events.
The Lipoprotein-Particle Data: Getting to the Heart of the Triad
Standard lipid panels report cholesterol concentrations, but atherogenic dyslipidemia is fundamentally a disorder of particle number and particle size. The most directly relevant retatrutide analysis, therefore, is not the routine panel but a post-hoc nuclear magnetic resonance (NMR) lipoprotein-particle study of the obesity phase 2 cohort, presented at the European Society of Cardiology Congress in 2024.9 This analysis interrogated exactly the variables that define the atherogenic phenotype, and its results are the closest the retatrutide literature comes to addressing the title’s question head-on.
At 48 weeks, the higher doses reduced triglycerides by up to about 40.6%, non-HDL-cholesterol by up to about 26.9%, apoB by up to about 24.2%, and apoC-III by up to about 38.0%.9 More revealing for this phenotype, the analysis reported reductions in triglyceride-rich lipoprotein particles across sizes, with the largest reductions in the large and medium particles, and reductions in total and small LDL particle number. HDL particle count fell slightly overall while average HDL particle size increased.9 Perhaps most striking, the NMR-derived lipoprotein insulin-resistance (LPIR) score — a composite of six lipoprotein subclass measures designed to capture precisely the insulin-resistant, atherogenic lipoprotein pattern — decreased by roughly 27.4% to 32.5% across the 4, 8, and 12 mg doses.9
Read against the pathophysiology laid out earlier, this is close to a textbook reversal of the atherogenic lipoprotein pattern: fewer triglyceride-rich particles, fewer small dense LDL particles, larger HDL, and a large drop in a validated composite index of the atherogenic, insulin-resistant lipoprotein state. If one wanted a drug-induced demonstration that triple agonism can shift human lipoprotein biology away from the atherogenic configuration, this is it. It is genuinely informative — and it is the strongest single reason the title’s premise is not absurd.
But the honest boundaries must be drawn just as sharply. This was a post-hoc analysis of a phase 2 trial not designed as a lipid study, in people without established atherogenic dyslipidemia as an entry criterion, over 48 weeks, with no clinical endpoints. Lipoprotein-particle changes are surrogate markers; they make a hypothesis compelling, but the history of cardiovascular medicine is littered with agents that improved lipid surrogates without improving outcomes, and a few that improved surrogates while worsening outcomes. The particle data inform the science; they do not license a therapeutic claim.
Weight-Mediated Versus Direct Effects: Disentangling the Mechanism
A central interpretive question — and one that determines how much of this is really about “retatrutide and lipids” as opposed to “weight loss and lipids” — is how much of the lipid improvement is a direct pharmacologic action and how much is the predictable consequence of losing 15 to 24% of body weight. Any substantial weight loss, by any means, improves atherogenic dyslipidemia, because it reduces the visceral adiposity and insulin resistance that drive hepatic VLDL overproduction in the first place.5 This is not a trivial confounder; it is arguably the dominant one.
Several lines of reasoning suggest that at least part of retatrutide’s lipid effect is more than a weight epiphenomenon, though none is conclusive. The liver-fat reductions in the MASLD cohort were large and, at the higher doses, out of proportion to what weight loss alone would typically produce, pointing to a direct hepatic action consistent with glucagon-receptor engagement.8 The preclinical glucagon-agonism data show triglyceride lowering that can occur through enhanced hepatic fat oxidation and suppressed lipogenesis, mechanisms that operate at the level of the hepatocyte rather than through adipose-tissue mass.6 And the early human pharmacology of the molecule, characterized in the first-in-human and phase 1b programs, established the dose-exposure relationships and glucagon-linked metabolic effects (including increased energy expenditure) that underlie these actions.10 The GLP-1-mediated reduction in hepatic VLDL-apoB output adds a further weight-independent route.7
Yet the counter-argument is strong and must be given its due. The lipid effects are dose-dependent, but so is the weight loss, so dose-dependence alone cannot separate the two. Without a formal mediation analysis — statistically partitioning the lipid change into the fraction explained by weight change and the fraction remaining after adjustment — we cannot confidently quantify the direct component in humans, and such analyses have not been the headline of the published phase 2 lipid reports. The intellectually honest position is that retatrutide almost certainly improves atherogenic lipids through a combination of substantial weight loss and a plausible direct hepatic action, with the direct component supported by mechanism and by the liver-fat data but not yet cleanly quantified in people. For readers interested in the parallel question of how these agents affect vascular and blood-pressure physiology, our discussion of how retatrutide influences cardiovascular risk factors takes up the broader cardiometabolic picture.
Why does the weight-versus-direct distinction matter for the title’s question? Because if the lipid benefit is almost entirely weight-mediated, then retatrutide “informs” dyslipidemia treatment in the same generic way any effective weight-loss intervention does, and the interesting biology lives in the weight loss, not the lipids. If, on the other hand, the glucagon arm contributes a genuine weight-independent hepatic lipid effect, then triple agonism represents a mechanistically novel lever on atherogenic dyslipidemia that is worth studying in its own right. The current evidence leans toward the latter being at least partly true, but leaves the magnitude unresolved.
How Retatrutide Compares With Lipid-Modifying Approaches
Placing retatrutide beside established and investigational lipid interventions clarifies both what its lipid signature resembles and where it sits in the therapeutic landscape. The comparison is instructive precisely because retatrutide has never been developed as a lipid drug; seeing which agents it resembles reveals what kind of lipid effect it actually has.
| Agent / class | Primary lipid action | Effect on atherogenic-dyslipidemia markers | Approved for dyslipidemia? |
|---|---|---|---|
| Statins | Upregulate LDL receptors; lower LDL-C | Large LDL-C and apoB reduction; modest TG effect; outcome-proven | Yes |
| Fibrates | PPAR-α activation | Large TG reduction, HDL rise; targets the triad; mixed outcome data | Yes (TG-focused) |
| Icosapent ethyl | Omega-3 (EPA) | TG reduction; outcome benefit in high-risk statin-treated patients | Yes (residual risk) |
| GLP-1 receptor agonists | Weight loss + hepatic effects | Moderate TG and modest apoB reduction; CV outcome benefit shown for some | No (approved for obesity/T2D) |
| Tirzepatide (GIP/GLP-1) | Weight loss + metabolic effects | TG, apoB, apoC-III reductions; larger than single GLP-1 | No (approved for obesity/T2D) |
| Retatrutide (GIP/GLP-1/glucagon) | Weight loss + direct hepatic lipid action | Large TG, non-HDL, apoB, apoC-III reductions; small dense LDL and LPIR down9 | No — investigational, not approved for any indication |
Two observations follow. First, retatrutide’s lipid profile is unmistakably that of a triglyceride-rich-lipoprotein and apoB agent — it behaves, on the lipid panel, more like a fibrate or a potent metabolic drug than like a statin. Its comparatively modest LDL-cholesterol effect is not a weakness in this context; it is confirmation that the drug acts on the atherogenic, insulin-resistant lipoprotein pattern rather than on the LDL-receptor axis.9 Second, and decisively, every agent in the “approved for dyslipidemia” column earned that status through dedicated lipid trials and, in most cases, cardiovascular outcomes trials. Retatrutide has neither. It sits in the same regulatory category as the GLP-1 and GIP/GLP-1 drugs — metabolic agents whose lipid effects are real, secondary, and not the basis of any lipid indication — except that retatrutide is not yet approved for anything at all.
The comparison also underscores a subtle but important point about what “good” looks like for this particular lipid disorder. A clinician treating classic hypercholesterolemia wants a large LDL-cholesterol reduction, and a statin delivers it. A clinician confronting atherogenic dyslipidemia in an obese, insulin-resistant patient wants something different: fewer triglyceride-rich remnants, a lower apoB particle count, a shift of LDL from small dense to large buoyant, and improvement in the insulin-resistant lipoprotein pattern that no LDL-cholesterol number captures. On that scorecard, retatrutide’s reported effects are well matched to the disorder — which is precisely why the research is interesting, and precisely why it must not be over-read. Matching a lipid profile is a reason to run the definitive trials, not a substitute for them.
A meta-analysis of retatrutide randomized trials has confirmed the direction and consistency of its cardiometabolic effects, including on lipids and blood pressure, while noting that the evidence base remains phase 2-dominated and that longer, outcome-oriented data are needed.11 That is a fair summary of where the comparison leaves us: retatrutide has a distinctive and favorable atherogenic-lipid signature, but it has not run the race that the approved lipid drugs have run.
The Evidence Gap: No Dyslipidemia Trial, No Outcomes Data
This section is deliberately blunt, because the gap it describes is the single most important fact for anyone tempted to read the lipid data as a therapeutic endorsement. There is, to date, no clinical trial in which retatrutide was administered specifically to treat atherogenic dyslipidemia, with atherogenic lipids as the primary endpoint, in a population selected for that lipid disorder. Every lipid result discussed above is a secondary or post-hoc finding from trials designed to study weight, glycemia, or liver fat.348
Just as importantly, there is no completed cardiovascular outcomes trial for retatrutide. We do not know whether the favorable lipoprotein changes it produces translate into fewer myocardial infarctions, strokes, or cardiovascular deaths. That is not a pedantic objection. The entire justification for treating atherogenic dyslipidemia is to prevent atherosclerotic events, and surrogate lipid improvements are accepted as a basis for treatment only for interventions whose outcome benefit has been demonstrated or can be confidently inferred from a large body of concordant evidence. Retatrutide has not reached that bar, and cannot until dedicated cardiovascular outcomes data exist.
The program is, however, advancing. Phase 3 development under the TRIUMPH (obesity) and TRANSCEND (diabetes) umbrellas is underway, and the first phase 3 diabetes trial, TRANSCEND-T2D-1, has reported: in 537 adults with type 2 diabetes, retatrutide lowered HbA1c by roughly 1.7 to 2.0% and produced weight loss up to about 16.8% at 40 weeks, with improvements again reported in non-HDL-cholesterol, triglycerides, and systolic blood pressure.12 This confirms the lipid direction in a larger, later-phase setting, but note what it still is: lipids as cardiometabolic secondary endpoints in a diabetes trial, not a dyslipidemia trial, and not an outcomes trial. Until a cardiovascular outcomes study reads out, the evidence for retatrutide in atherogenic dyslipidemia will remain mechanistically rich and clinically suggestive but formally unproven for that purpose.
The correct scientific posture toward the title’s question, then, is this: retatrutide research informs our understanding of how triple GIP/GLP-1/glucagon agonism reshapes atherogenic lipoprotein metabolism, and it generates a serious hypothesis that such agonism could benefit patients with this lipid phenotype. It does not, and given the current data cannot, tell us that retatrutide should be used to treat atherogenic dyslipidemia.
Dosing and Handling in a Research Context
Because retatrutide is investigational, any discussion of dosing must be framed strictly as a description of what was studied in trials and how research material is handled, not as guidance for use. Nothing in this section is a recommendation, and self-directed use of a non-approved peptide outside a regulated trial carries real and unquantified risk.
In the clinical trials, retatrutide was given once weekly by subcutaneous injection, with a stepwise titration schedule intended to mitigate the gastrointestinal side effects characteristic of the incretin class. Studied maintenance doses spanned roughly 1 mg to 12 mg weekly, with the largest metabolic and lipid effects observed at 8 and 12 mg after titration from lower starting doses; the phase 3 diabetes program titrated from 2 mg toward maintenance doses of 4, 9, and 12 mg.12 The titration is not incidental — the tolerability and, arguably, the safety of the drug depend on the gradual dose escalation, and the lipid and metabolic benefits accrue at the higher maintenance doses reached only after that escalation.
Retatrutide supplied for laboratory research is typically a lyophilized (freeze-dried) powder requiring reconstitution with a sterile diluent before any in-vitro or preclinical use. Standard research-peptide practice — directing diluent gently against the vial wall rather than onto the powder, swirling rather than shaking to avoid shearing the peptide, protecting the material from light, heat, and repeated freeze-thaw cycles, and observing aseptic technique — applies here as it does to any peptide of this class. Detailed, education-only handling and stability considerations for this specific compound are compiled in our guide to the critical handling protocols for retatrutide research studies, and general reconstitution arithmetic is covered in our peptide research glossary.
It bears repeating that meticulous handling does nothing to change the evidence question. A perfectly reconstituted, high-purity vial of retatrutide is still a compound with no approved indication and no dyslipidemia trial behind it. Good technique preserves whatever activity the molecule has; it does not create clinical validation where none exists. A further, non-trivial concern with material obtained outside regulated channels is quality: peptides sold as “research chemicals” vary in purity, and impurities, incorrect mass, or endotoxin contamination introduce hazards that are entirely separate from the pharmacology of retatrutide itself.
Safety and Tolerability
Any consideration of retatrutide for a chronic, largely asymptomatic condition like dyslipidemia has to weigh tolerability heavily, because the threshold for acceptable side effects is lower when treating a risk factor than when treating an acute illness. The safety data, like the efficacy data, come from phase 2 and now early phase 3 trials of limited duration.
The dominant adverse effects are gastrointestinal — nausea, vomiting, diarrhea, and constipation — overwhelmingly during dose escalation, consistent with the GLP-1 and GIP/GLP-1 classes; these are generally mild to moderate and diminish with time and slower titration.34 The phase 3 diabetes trial reported treatment-emergent adverse events typical of incretin therapy, with adverse-event discontinuations in the low single-digit percentages and a small incidence of dysesthesia.12 A mechanism-specific consideration is the glucagon component: because glucagon can raise glucose and heart rate, trials monitored these closely; the calibrated, sub-native glucagon potency appears to allow the metabolic benefits without net glycemic worsening — indeed glycemia improved — but modest increases in heart rate, seen across incretin agents, warrant attention, particularly in a cardiovascular-risk population.
Several caveats specific to a dyslipidemia framing follow. First, the safety database reflects durations of roughly 24 to 48 weeks in phase 2 and up to about 40 weeks in the first phase 3 readout; dyslipidemia is a lifelong condition, and long-term safety of retatrutide is not yet characterized. Second, the trial populations were selected (obesity, type 2 diabetes, MASLD) and may not represent the broader dyslipidemic population, including individuals who are not substantially overweight. Third, and most importantly, an acceptable short-term tolerability profile is not evidence of clinical benefit; a drug can be well tolerated and still fail to improve outcomes. Absence of major short-term safety signals and absence of demonstrated dyslipidemia-outcome benefit coexist here, and both should be stated plainly.
Regulatory Status
Precision matters here, because the gap between a drug’s scientific promise and its regulatory standing is exactly where honest writing earns its keep.
As of mid-2026, retatrutide is an investigational agent that is not approved by the FDA, the EMA, or any comparable major regulator for any indication. It is not approved for obesity, not for type 2 diabetes, not for MASLD, and not — the point of this article — for atherogenic dyslipidemia or cardiovascular risk reduction. Its phase 3 program is ongoing, and the first phase 3 diabetes trial has reported positive results on glycemia and weight with supportive lipid and blood-pressure secondary endpoints,12 but positive phase 3 data are a step toward possible approval, not approval itself, and the anticipated first indications will be obesity and type 2 diabetes rather than dyslipidemia.
There is, correspondingly, no approved indication for retatrutide that could be extended to atherogenic dyslipidemia, and no regulatory body has evaluated it for that use. Any lipid benefit it ultimately delivers to patients would, in the foreseeable pathway, arrive as a secondary consequence of treating obesity or diabetes, not as a standalone dyslipidemia indication — unless a dedicated program with lipid and cardiovascular outcome endpoints were undertaken, which as of this writing has not been announced. The material sold online as “research retatrutide” is not a regulated medicine, is not quality-assured to pharmaceutical standards, and its use in humans outside an authorized clinical trial is neither sanctioned nor advisable.
The regulatory synthesis is therefore straightforward and worth stating without hedging: retatrutide is a promising investigational triple agonist whose phase 2 and early phase 3 data show robust, favorable effects on the atherogenic lipoprotein pattern, and which is not approved to treat dyslipidemia, cardiovascular disease, or anything else. The proper path for exploring its lipid potential is formal clinical investigation under regulatory oversight, including the cardiovascular outcomes trials that alone can convert an attractive lipid surrogate into a justified therapy. Readers tracking how the incretin-glucagon class is reshaping metabolic medicine more broadly may find our overview of GLP-1 signaling mechanisms in arterial stiffness a useful companion.
Frequently Asked Questions
Is retatrutide approved to treat atherogenic dyslipidemia?
No. Retatrutide is an investigational triple GIP/GLP-1/glucagon receptor agonist that, as of mid-2026, is not approved by the FDA, EMA, or any comparable regulator for any indication, including dyslipidemia or cardiovascular risk reduction. Its favorable effects on triglycerides, non-HDL-cholesterol, apolipoprotein B, and other atherogenic markers come from phase 2 trials (and early phase 3 secondary endpoints) in obesity, type 2 diabetes, and liver disease — not from any trial designed to treat dyslipidemia.3412
What lipid changes did retatrutide actually produce in trials?
Across the phase 2 obesity and diabetes trials, higher doses reduced triglycerides by up to roughly 40%, and a post-hoc lipoprotein analysis of the obesity cohort reported reductions in non-HDL-cholesterol (up to ~27%), apolipoprotein B (up to ~24%), and apolipoprotein C-III (up to ~38%), along with fewer small dense LDL particles, larger HDL particles, and a large drop in a lipoprotein insulin-resistance score.39 LDL-cholesterol changed less and less consistently — a pattern typical of a drug acting on triglyceride-rich lipoproteins rather than the LDL-receptor pathway.
Why would a glucagon-receptor agonist help with lipids?
Glucagon acts directly on the liver to increase fatty-acid oxidation and suppress the manufacture of new fat, which reduces the hepatic triglyceride pool available for export as VLDL. In animal studies, glucagon-receptor agonism lowers plasma and liver triglycerides while antagonism raises them.6 Because hepatic VLDL overproduction is the upstream driver of atherogenic dyslipidemia, a calibrated glucagon action — combined with GLP-1’s effect on hepatic VLDL output — is a mechanistically coherent reason retatrutide moves these lipids.78
Are retatrutide’s lipid benefits just from weight loss?
Partly, and perhaps largely. Losing 15 to 24% of body weight improves atherogenic dyslipidemia by any means, because it reduces the insulin resistance that drives hepatic VLDL overproduction.5 However, the very large liver-fat reductions in the MASLD trial and the preclinical glucagon-agonism data suggest a direct hepatic component beyond weight alone.68 Without a formal mediation analysis, the exact split between weight-mediated and direct effects in humans remains unquantified.
Does improving these lipid markers mean retatrutide prevents heart attacks?
Not established. Retatrutide has no completed cardiovascular outcomes trial, so we do not know whether its favorable lipoprotein changes translate into fewer cardiovascular events. Lipid and lipoprotein-particle measures are surrogate markers; the history of cardiovascular medicine includes agents that improved lipid surrogates without improving, or even while worsening, outcomes. Outcome data would be required before any preventive claim.
How does retatrutide compare with tirzepatide or a GLP-1 drug for lipids?
Retatrutide adds a third receptor — glucagon — that directly targets the liver, the organ whose VLDL overproduction underlies atherogenic dyslipidemia. Its reported reductions in triglycerides, apoB, and apoC-III are in the same favorable direction as GLP-1 and GIP/GLP-1 agonists and, at higher doses, appear at least as large; the tuned glucagon component is the mechanistic differentiator.19 That said, like those drugs, retatrutide is not approved for dyslipidemia, and none of these agents earned lipid indications through dedicated lipid trials.
Was retatrutide studied in people who have atherogenic dyslipidemia specifically?
No. The trials enrolled people with obesity, type 2 diabetes, or MASLD — populations in whom atherogenic dyslipidemia is common but which were not selected for that lipid disorder, and in which lipids were secondary rather than primary endpoints.348 There is no dedicated dyslipidemia trial of retatrutide with atherogenic lipids as the primary outcome.
What is the current stage of retatrutide development?
It is in phase 3 development, with the first phase 3 type 2 diabetes trial (TRANSCEND-T2D-1) reporting strong glycemic and weight results and supportive lipid and blood-pressure secondary endpoints.12 Phase 3 obesity trials are ongoing. Positive phase 3 data move a drug toward possible approval but are not approval, and the anticipated first indications are obesity and type 2 diabetes, not dyslipidemia.
Is it safe to use research retatrutide to improve my cholesterol?
No responsible answer supports that. Retatrutide is not an approved medicine; material sold for research is not quality-assured to pharmaceutical standards; long-term safety is uncharacterized; and there is no evidence that using it improves cardiovascular outcomes. Established, outcome-proven options for atherogenic dyslipidemia — lifestyle change, statins, and where indicated triglyceride-directed therapies — should be managed with a qualified clinician. This article is educational and is not medical advice.
References
- Coskun T, Urva S, Roell WC, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: from discovery to clinical proof of concept. Cell Metab. 2022;34(9):1234-1247.e9. PMID: 35985340. https://pubmed.ncbi.nlm.nih.gov/35985340/
- Manjunath CN, Rawal JR, Irani PM, Madhu K. Atherogenic dyslipidemia. Indian J Endocrinol Metab. 2013;17(6):969-976. PMID: 24381869. PMCID: PMC3872713. https://pmc.ncbi.nlm.nih.gov/articles/PMC3872713/
- Jastreboff AM, Kaplan LM, Frías JP, et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity — A Phase 2 Trial. N Engl J Med. 2023;389(6):514-526. PMID: 37366315. https://pubmed.ncbi.nlm.nih.gov/37366315/
- Rosenstock J, Frías J, Jastreboff AM, et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: a randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet. 2023;402(10401):529-544. PMID: 37385280. https://pubmed.ncbi.nlm.nih.gov/37385280/
- Hirano T. Pathophysiology of Diabetic Dyslipidemia. J Atheroscler Thromb. 2018;25(9):771-782. PMID: 29998913. PMCID: PMC6143775. https://pmc.ncbi.nlm.nih.gov/articles/PMC6143775/
- Galsgaard KD, Elmelund E, Johansen CD, et al. Glucagon receptor antagonism impairs and glucagon receptor agonism enhances triglycerides metabolism in mice. Mol Metab. 2022;66:101639. PMID: 36400402. PMCID: PMC9706156. https://pmc.ncbi.nlm.nih.gov/articles/PMC9706156/
- Taher J, Baker CL, Cuizon C, et al. GLP-1 receptor agonism ameliorates hepatic VLDL overproduction and de novo lipogenesis in insulin resistance. Mol Metab. 2014;3(9):823-833. PMID: 25506548. PMCID: PMC4264039. https://pmc.ncbi.nlm.nih.gov/articles/PMC4264039/
- Sanyal AJ, Kaplan LM, Frias JP, et al. Triple hormone receptor agonist retatrutide for metabolic dysfunction-associated steatotic liver disease: a randomized phase 2a trial. Nat Med. 2024;30(7):2037-2048. PMID: 38858523. PMCID: PMC11271400. https://pmc.ncbi.nlm.nih.gov/articles/PMC11271400/
- Nicholls SJ, Pirro V, Lin Y, et al. Triple-hormone receptor agonist retatrutide significantly improves lipoprotein and apolipoprotein profiles in participants with obesity or overweight. Eur Heart J. 2024;45(Suppl 1):ehae666.1501. (Abstract presented at the European Society of Cardiology Congress 2024; post-hoc NMR lipoprotein-particle analysis of the phase 2 obesity trial.) https://academic.oup.com/eurheartj/article/45/Supplement_1/ehae666.1501/7836502
- Urva S, Coskun T, Loh MT, et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. Lancet. 2022;400(10366):1869-1881. PMID: 36354040. https://pubmed.ncbi.nlm.nih.gov/36354040/
- Abouelmagd AA, Abdelrehim AM, Bashir MN, et al. Efficacy and safety of retatrutide, a novel GLP-1, GIP, and glucagon receptor agonist for obesity treatment: a systematic review and meta-analysis of randomized controlled trials. Proc (Bayl Univ Med Cent). 2025;38(3):291-303. PMID: 40291085. PMCID: PMC12026077. https://pmc.ncbi.nlm.nih.gov/articles/PMC12026077/
- Frias JP, Davies MJ, Rosenstock J, et al. Efficacy and safety of retatrutide, a GIP, GLP-1, and glucagon receptor agonist, in people with type 2 diabetes and inadequate glycaemic control with diet and exercise (TRANSCEND-T2D-1): a double-blind, randomised, phase 3 trial. Lancet. 2026. PMID: 42250575. https://pubmed.ncbi.nlm.nih.gov/42250575/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. Retatrutide is an investigational triple GIP/GLP-1/glucagon receptor agonist that is not approved by the FDA, EMA, or any comparable regulator for the treatment, cure, or prevention of atherogenic dyslipidemia, cardiovascular disease, obesity, diabetes, or any other condition. Its effects on atherogenic lipoproteins derive from phase 2 and early phase 3 trials in which lipids were secondary or exploratory endpoints; no dedicated dyslipidemia trial and no completed cardiovascular outcomes trial exist, and retatrutide has not been shown to reduce cardiovascular events. Nothing here is medical advice or a recommendation for human use. Material sold as “research retatrutide” is not a quality-assured medicine, and use outside an authorized clinical trial is not advisable. Readers should consult qualified healthcare professionals and applicable regulations before making any decisions about lipid management or any therapy.