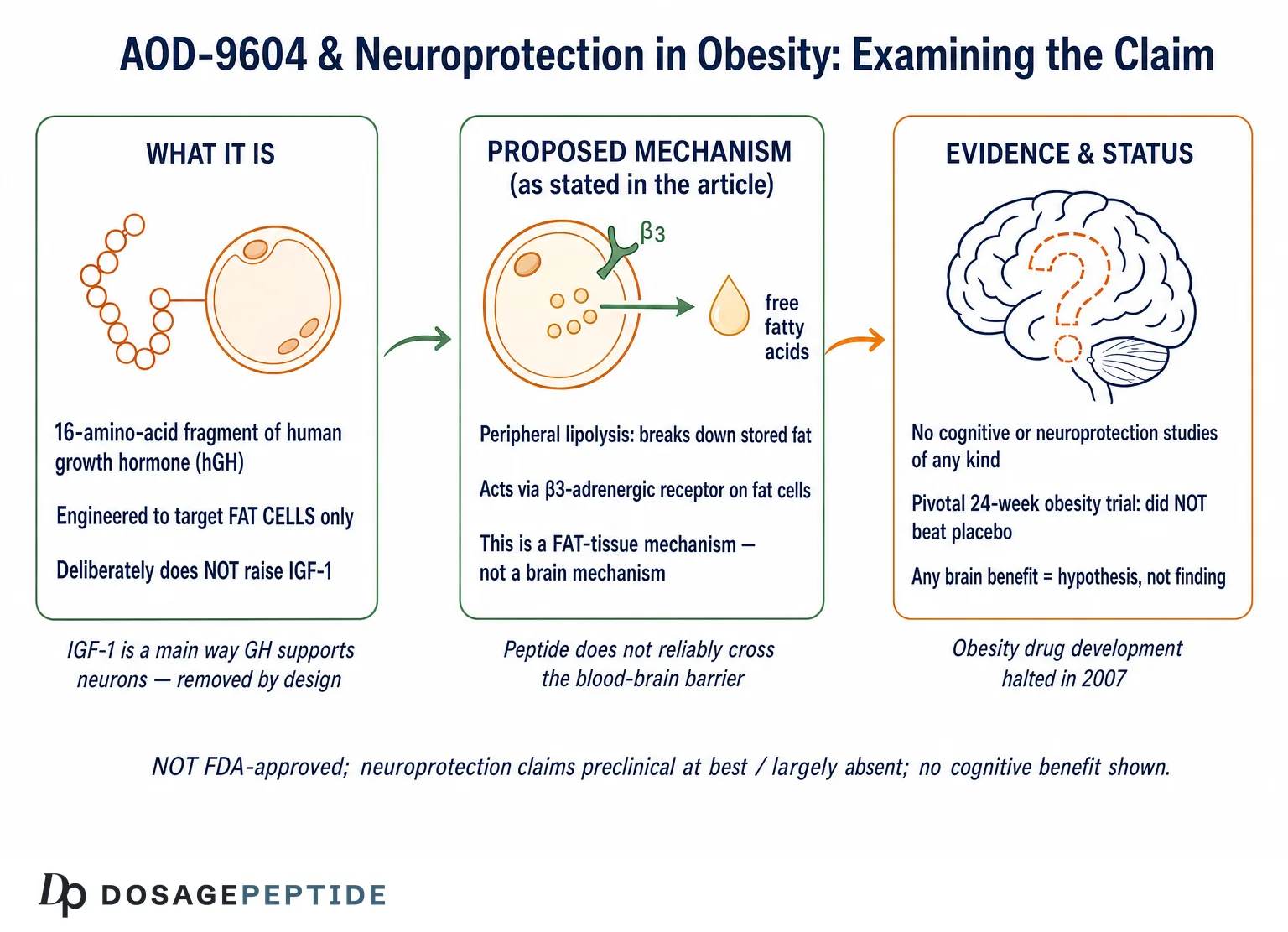
The question in the title assumes something that deserves scrutiny before we accept it: that AOD-9604 has “neuroprotective effects” in obese phenotypes, and that our task is merely to explain the molecular mechanisms behind them. That framing runs several steps ahead of the evidence. It presupposes (1) that AOD-9604 protects neurons, (2) that this protection is established in the context of obesity, and (3) that a well-characterized mechanism exists to be described. None of those three claims survives contact with the primary literature. AOD-9604 is a peripheral, fat-cell-targeting fragment of human growth hormone that was engineered specifically to act outside the endocrine and central nervous system pathways of the parent hormone.1 There is no controlled study — in humans, in animals, or in neuronal cell culture — demonstrating that it protects brain tissue.
So this article does not affirm the premise. It examines it. The honest posture toward the phrase “molecular mechanisms driving AOD-9604 neuroprotective effects” is to treat it as an open, largely unstudied research question and to map carefully what is known, what is plausible but untested, and what is simply absent. What we do have is a body of metabolic and obesity research on AOD-9604 — roughly six human clinical trials enrolling more than 900 participants, according to the sponsor’s development-program summary, terminated in 2007 when the pivotal trial failed to beat placebo.8 Separately, there is a rich and growing neuroscience literature on how obesity injures the brain.10 The title implicitly bridges these two bodies of work, but that bridge has never been built with data.
This piece is written for researchers and scientifically literate readers who want an accurate account rather than a marketing one. We will cover what AOD-9604 is and why it was designed the way it was, its actual molecular mechanism, the blood-brain-barrier problem that constrains any central action, the real mechanisms by which obesity harms neurons, the indirect “lose-weight-to-protect-the-brain” hypothesis and why the compound’s own weight-loss data undercut it, the complete absence of direct neuroprotection evidence, the human trial record, comparisons with interventions that have been studied in the obese brain, research methodology, safety, laboratory handling, and regulatory status. The guiding principle throughout is restraint: AOD-9604 is not approved for any indication, and nothing here should be read as suggesting it treats, prevents, or reverses cognitive decline, neurodegeneration, or any neurological disease.
What AOD-9604 Is and Why It Was Engineered
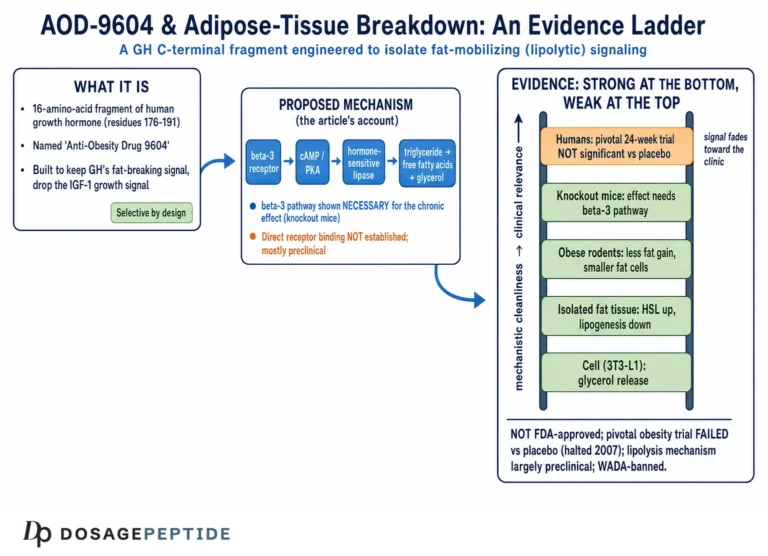
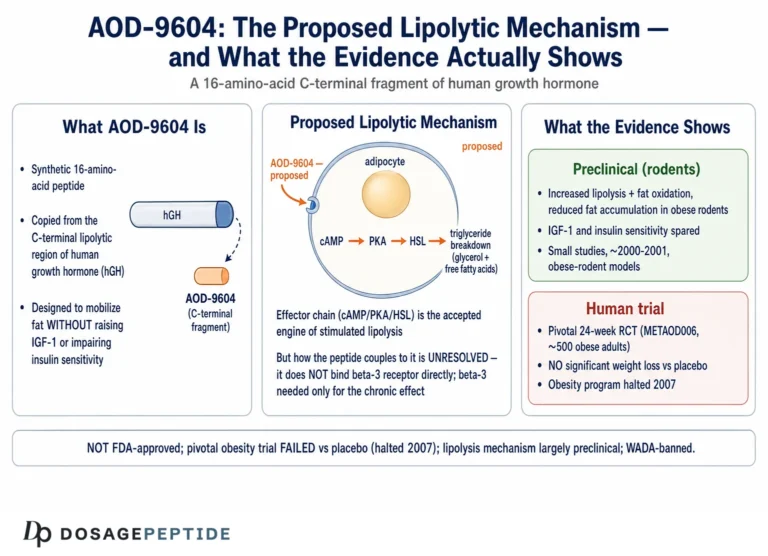
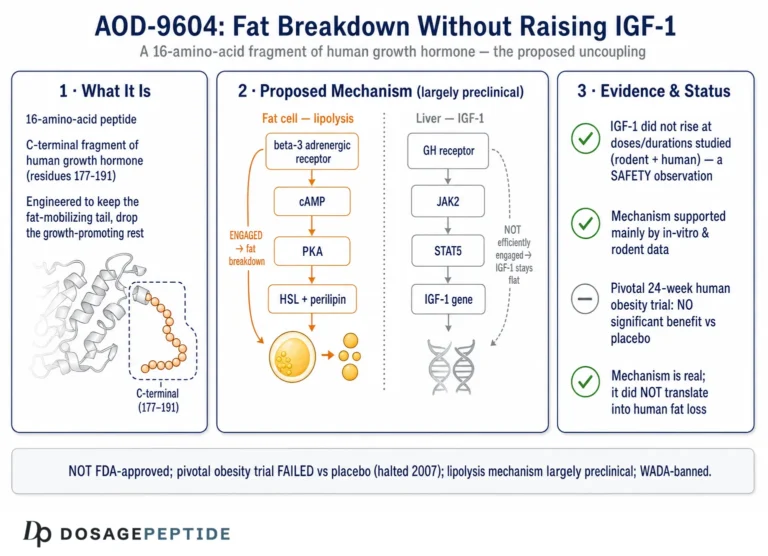
AOD-9604 is a synthetic 16-amino-acid peptide. The name abbreviates “Anti-Obesity Drug 9604,” which tells you the intent behind its creation more plainly than any mechanism diagram could. Its sequence corresponds to residues 176–191 of the C-terminal region of human growth hormone (hGH), with one deliberate modification: a tyrosine residue is added at the N-terminus, a change made to improve stability and handling of the fragment.1 Two cysteine residues form a disulfide bridge that reproduces the small loop found in the parent hormone’s lipolytic domain. This is a small, structurally constrained peptide built to mimic one narrow slice of growth-hormone biology.
The compound emerged from work at Monash University in Melbourne, Australia, and was developed commercially by Metabolic Pharmaceuticals Limited. The scientific logic was elegant and worth restating because it bears directly on the neuroprotection question. Full-length growth hormone does many things: it drives longitudinal bone growth, raises circulating insulin-like growth factor 1 (IGF-1), alters glucose handling, and, separately, promotes the breakdown of stored fat. Researchers had localized much of the fat-mobilizing (lipolytic) activity to the C-terminal portion of the molecule. The hope was that a short peptide reproducing only that region could deliver a fat-burning signal while leaving the other, potentially problematic, growth-hormone effects behind — particularly the rise in IGF-1 and the deterioration in glucose tolerance associated with chronic GH exposure.12
Early metabolic studies reported that AOD-9604 reproduced the lipolytic and fat-oxidizing actions of hGH in adipose tissue and in obese-rodent models without raising IGF-1 or impairing glucose tolerance.13 That selectivity became the compound’s defining property. It is also the property that makes the neuroprotection premise suspect. IGF-1 is one of the most important trophic and neuroprotective signals in the brain; a great deal of growth hormone’s plausible central benefit flows through the GH/IGF-1 axis. AOD-9604 was engineered to exclude that axis. So the very design choice that defines the molecule strips away the growth-hormone pathway most associated with neuronal support.
A useful discipline when reasoning about AOD-9604 is to hold three distinct things apart, because popular writing constantly blurs them. First, the parent hormone: full-length hGH, a 191-amino-acid protein with broad endocrine and central effects, including actions relayed through IGF-1. Second, the lipolytic domain: the C-terminal region thought to carry the fat-mobilizing signal. Third, the engineered fragment: AOD-9604 itself, a stabilized 16-residue reproduction of that domain, deliberately uncoupled from IGF-1. Each step narrows the biology. When someone reasons from “growth hormone influences the brain” to “this growth-hormone fragment protects the brain,” they are collapsing all three levels back into one and ignoring the deliberate narrowing that is the entire point of the molecule. That collapse is the single most common error in the claims surrounding this compound, and it recurs whether the alleged benefit is muscle growth, wound healing, or neuroprotection. Readers can see the same reasoning pattern examined for a different tissue in the discussion of whether AOD-9604 could aid recovery from muscle wasting in cachexia or sarcopenia, where the same IGF-1-sparing design similarly undercuts the claimed benefit.
Historically, the obesity program was the whole story for years. Metabolic Pharmaceuticals ran a series of human trials through the mid-2000s, and when the pivotal study underperformed, development as an anti-obesity drug was halted in 2007.8 The molecule then reappeared in two very different guises: as a self-affirmed “generally recognized as safe” ingredient marketed for metabolic-health supplements, and as an experimental intra-articular agent for osteoarthritis in animal models.56 None of these directions involved the brain.
The Molecular Mechanism: A Peripheral Lipolytic Signal
The proposed mechanism of AOD-9604 centers on adipocytes, the cells that store fat. In simplified terms, the peptide is thought to reproduce the lipolytic signaling of growth hormone’s C-terminal domain, promoting the breakdown of stored triglycerides into free fatty acids (lipolysis) while dampening the conversion of substrate into new fat (lipogenesis).1 The net effect in rodent models was reduced fat accumulation and increased fat oxidation, without the IGF-1 elevation that accompanies full growth-hormone administration.
The most frequently repeated mechanistic claim is that AOD-9604 works through the beta-3 adrenergic receptor (β3-AR) on fat cells to raise cyclic AMP and activate hormone-sensitive lipase. This claim deserves to be handled with care, because it is both the best-supported piece of AOD-9604 mechanism and the piece most often over-simplified. The strongest primary evidence comes from a 2001 study in which chronic treatment with hGH and with AOD-9604 reduced body weight and body fat in obese mice, with effects correlating with increased expression of β3-AR, whereas β3-AR knockout mice were unresponsive to the compound’s lipolytic effect.2 That knockout result implicates the β3-adrenergic pathway in the compound’s chronic metabolic action in fat tissue. Notably, in the same work some acute increases in energy expenditure and fat oxidation persisted in knockout animals, suggesting at least one β3-independent component to the acute response.2
What matters for this article is where that mechanism is located. The β3-adrenergic receptor is expressed predominantly on peripheral adipose tissue — white and brown fat — not on the neuronal populations whose survival defines neuroprotection. The mechanism, as characterized, is a story about fat cells and the sympathetic-adrenergic control of lipolysis. Nothing in the primary AOD-9604 mechanism literature describes an action on neurons, glia, synapses, the blood-brain barrier, amyloid processing, tau, oxidative-stress defenses in brain tissue, or any of the molecular systems that neuroprotection research actually measures. The honest one-sentence summary is that AOD-9604 has a reasonably characterized peripheral, fat-directed mechanism anchored to at least one solid knockout study, and essentially no characterized central-nervous-system mechanism at all.
This is the crux of the title’s problem. To describe “molecular mechanisms driving neuroprotective effects,” there must first be a demonstrated neuroprotective effect and then a mechanism connecting the compound to neurons. AOD-9604 supplies neither. Any mechanistic account of central protection would have to be constructed by analogy or speculation — borrowing the reputation of full-length growth hormone or of IGF-1, precisely the signals the molecule was built to avoid. For readers surveying how growth-hormone-related research peptides are distinguished by their metabolic versus trophic profiles, the site’s dosage index catalogs these compounds by their intended actions rather than by aspirational claims.
The Blood-Brain-Barrier Problem
There is a physical, not merely evidentiary, obstacle standing between AOD-9604 and any direct neuroprotective action: the blood-brain barrier (BBB). Peptides do not cross this barrier freely. The BBB is a tightly regulated interface of endothelial cells joined by tight junctions, supported by pericytes and astrocytic end-feet, that excludes most large, hydrophilic, charged molecules unless there is a specific transport system to carry them across. A 16-amino-acid peptide with a disulfide-constrained loop, designed for peripheral fat action, has no established central transport route and no evidence of meaningful brain penetration.
This is not an incidental limitation; in one sense it was a design goal. Part of the appeal of AOD-9604 as an obesity candidate was that its fat-mobilizing action appeared to be peripheral and did not depend on central appetite suppression. Unlike agents that act on hypothalamic feeding circuits, AOD-9604 was characterized as working directly on adipose tissue, without the central nervous system effects on eating behavior that define appetite-acting drugs. A compound whose selling point is that it acts on fat without reaching central feeding centers is, by that same logic, a poor candidate to exert direct protective effects on cortical or hippocampal neurons deep behind the BBB.
The consequence for mechanism is stark. If a molecule does not reach neurons in appreciable concentration, then any neuroprotective effect it might have cannot be direct — it cannot be receptor binding on neurons, direct modulation of neuronal survival pathways, or local scavenging of reactive species in brain parenchyma. It would have to be indirect, mediated entirely through peripheral changes (fat mass, circulating lipids, systemic inflammation, insulin sensitivity) that then influence the brain from outside. That distinction — direct central action versus indirect peripheral-to-central signaling — is the whole game when evaluating the neuroprotection premise, and the BBB pushes AOD-9604 firmly into the “indirect, if anything” category. The next sections examine whether even that indirect route is supported.
How Obesity Injures the Brain — the Mechanisms the Title Invokes
To judge whether AOD-9604 could plausibly protect the obese brain, one must be precise about how obesity harms the brain in the first place, because the title’s phrase “neuroprotective effects in obese phenotypes” borrows the credibility of a genuine and well-studied field — obesity-associated cognitive decline — and attaches it to a compound that field has never tested.
The dominant, evidence-supported model is that obesity and high-fat feeding produce chronic low-grade systemic inflammation, elevated circulating free fatty acids, insulin resistance, and vascular dysfunction, and that these peripheral changes are transmitted into the brain where they drive neuroinflammation.10 Circulating cytokines, saturated fatty acids, and immune signals reach the brain especially at the hypothalamus, where saturated fatty acids and lipopolysaccharide can stimulate toll-like receptor 4 (TLR4) on microglia and initiate an inflammatory cascade.10 Activated microglia proliferate and shift toward a pro-inflammatory phenotype, releasing tumor necrosis factor, interleukin-1β, and interleukin-6. In the hippocampus — a region central to learning and memory — this neuroinflammation is associated with impaired long-term potentiation, reduced neurogenesis, synaptic remodeling, and, in animal models, measurable deficits in hippocampus-dependent memory.1011
Mechanistic work has drilled into specific molecular axes within this cascade. For example, a microglial fatty-acid-binding-protein-4 (FABP4)–UCP2 axis has been implicated in modulating neuroinflammation and cognitive decline in obese mice, illustrating how lipid-handling machinery inside brain immune cells links a fatty diet to cognitive outcomes.12 Beyond inflammation, obesity contributes to cerebral cognitive risk through vascular changes, blood-brain-barrier disruption, altered brain insulin signaling, oxidative stress, mitochondrial dysfunction, and, over longer horizons, associations with amyloid-β pathology and increased dementia risk.11
Notice the shape of this biology. The drivers are inflammatory (cytokines, TLR4, microglial activation), vascular, and insulin-related. The therapeutic logic that follows — if one wanted to protect the obese brain — would be to dampen neuroinflammation, restore insulin sensitivity, repair vascular and BBB integrity, or reduce the peripheral inflammatory load that feeds the whole process. A purely lipolytic signal that mobilizes stored fat into circulating free fatty acids is not obviously aligned with any of these targets. Indeed, acutely raising circulating free fatty acids is, in the obesity-brain literature, part of the problem: free fatty acids are among the mediators that stimulate TLR4 and promote central inflammation.10 A compound whose signature action is to accelerate fat breakdown into free fatty acids is, at least on its face, poorly matched to a syndrome in which circulating lipids help drive the injury. This does not prove AOD-9604 would worsen brain outcomes — no one has measured it — but it does mean the mechanistic case for benefit is weak and would have to rest on net long-term reductions in adiposity outweighing any acute lipid effect.
The Indirect Neuroprotection Hypothesis and Why the Weight Data Undercut It
Since a direct central action is implausible on BBB grounds, the only remotely defensible version of the neuroprotection premise is indirect: perhaps by reducing body fat, AOD-9604 would lower systemic inflammation, improve insulin sensitivity, and thereby reduce the peripheral drivers of obesity-related brain injury — an indirect neuroprotection by way of metabolic improvement. It is a coherent hypothesis in principle. Sustained weight loss and improved metabolic health are genuinely associated with better cognitive trajectories in obese populations, and interventions that reduce adiposity and inflammation are a rational strategy for the obese brain.11
The trouble is that this indirect route depends entirely on AOD-9604 actually producing meaningful, durable fat loss and metabolic improvement in humans — and that is exactly what its clinical program failed to demonstrate. The chain of reasoning “peptide → large fat loss → less systemic inflammation → less neuroinflammation → protected neurons” breaks at its very first link. In the pivotal human trial, AOD-9604 did not separate from placebo on weight loss to a statistically significant degree, and development was halted.8 If the compound cannot reliably deliver the metabolic improvement on which the entire indirect hypothesis rests, the downstream neuroprotective inference collapses. You cannot claim a brain benefit that is contingent on weight loss when the weight loss itself was not robustly demonstrated.
It is worth contrasting this with agents whose metabolic effects are established. Incretin-based therapies such as tirzepatide and the investigational triple agonist retatrutide produce large, well-documented weight loss in randomized trials, and there is active, legitimate research interest in whether their metabolic and anti-inflammatory effects translate into neurological benefit — a question still being investigated rather than settled. Readers can see how the evidence bar is set for compounds with real efficacy in the discussions of retatrutide as a triple-receptor agonist. The point of the contrast is not to elevate those agents as proven neuroprotectants — they are not — but to show that even for compounds with unambiguous weight-loss efficacy, the leap to neuroprotection remains an open research question. For AOD-9604, which lacks the underlying efficacy, that leap is not merely unproven; it is unsupported at the foundation.
A parallel confusion is worth flagging because it recurs across the growth-hormone-peptide field: the assumption that anything improving metabolic health must improve brain health, as if body and brain moved on a single dial. They do not. Metabolic improvement can support cognition, but the relationship is probabilistic, population-level, and dependent on the magnitude and durability of the metabolic change. A marginal, non-significant weight effect does not reliably move the inflammatory and vascular processes that injure the obese brain. The dissociation matters, and it is why the neuroprotection question cannot be answered by gesturing at a fat-loss mechanism.
Direct Neuroprotection Evidence for AOD-9604: The Gap
This section is brief because the honest answer is brief: there is no direct evidence — clinical or controlled preclinical — that AOD-9604 protects neurons or improves cognition in obese phenotypes or in any other setting. A search of the primary literature returns no clinical cognitive trials, no dementia or mild-cognitive-impairment studies, no controlled animal neurodegeneration models (high-fat-diet cognitive-decline models, Alzheimer’s-model mice, ischemia/stroke models, or otherwise) in which AOD-9604 was tested as a neuroprotective intervention, and no neuronal or glial cell-culture work establishing an effect on neuronal survival, oxidative-stress defenses, or inflammatory signaling.
It is worth being candid about how the contrary impression arises. Because AOD-9604 is a growth-hormone fragment, and because obesity is genuinely linked to cognitive decline, marketing and secondary sources sometimes stitch the two facts together into an implied benefit — “a growth-hormone peptide that helps the obese brain.” This is a category error built from two true statements and one false bridge. Growth hormone’s central effects flow substantially through IGF-1; AOD-9604 was repeatedly reported not to elevate IGF-1.13 The compound therefore lacks the very signal through which the parent hormone might plausibly support neurons, and it cannot reliably reach the brain to act by any other route. The neuroprotection claim is not weakly supported; on the specific question of AOD-9604 protecting neurons in obese phenotypes, the evidence level is zero.
The peptide’s only well-documented regenerative signal outside fat metabolism is in cartilage, not brain. In a collagenase-induced knee osteoarthritis model in rabbits, weekly intra-articular injections of AOD-9604 (0.25 mg), alone or combined with hyaluronic acid, improved gross and histopathological cartilage scores relative to saline controls.5 This is a genuine, peer-reviewed preclinical finding — but it concerns joint cartilage under direct local injection, an entirely different tissue, delivery route, and question from neuronal protection behind the BBB. It cannot be borrowed to support a brain claim. A separate line of speculation about tissue repair and wound healing is examined in the discussion of whether AOD-9604 could enhance healing in diabetic foot ulcers and wound repair, and it, too, rests on preclinical inference rather than demonstrated clinical benefit. For a companion treatment of the same neuroprotection question framed around obesity-related cognitive decline specifically, see the related analysis of what research links AOD-9604 to neuroprotection in obesity-related cognitive decline, which reaches the same conclusion: the connection is hypothesized, not demonstrated.
What the Human Obesity Trials Actually Showed
Because AOD-9604’s clinical record lives almost entirely in obesity, that record is the fairest way to gauge the compound’s demonstrated capabilities — and, by extension, the plausibility of any indirect neuroprotection. The short version: it was tested seriously, in humans, at scale, and it did not deliver a robust metabolic effect.
Across its development, AOD-9604 was studied in roughly six human clinical trials enrolling more than 900 participants in total, a tally that derives from the Metabolic Pharmaceuticals development-program summary rather than from any single published paper.8 The centerpiece early study was a 12-week Phase 2 evaluation in obese adults (designated METAOD005) using once-daily oral administration across several dose arms. Its data were encouraging enough to generate optimistic press: in that 12-week analysis, peptide-treated groups lost on the order of 1.8–2.6 kg more than placebo depending on the arm, with the 1 mg dose reported as the best performer.8
The problem was what happened at the endpoints that mattered. In the pivotal, longer, more rigorously controlled 24-week evaluation — METAOD006, a randomized, double-blind, placebo-controlled, multicenter study, and the single published RCT on the compound — the weight-loss difference between AOD-9604 and placebo did not reach statistical significance at the primary and key secondary endpoints.8 The early signal from the shorter study faded against a background of intensive diet-and-exercise intervention; the peptide added no detectable benefit over lifestyle alone. Development as an obesity drug was terminated in 2007.
| Aspect | What the obesity program showed |
|---|---|
| Human trials | ~6 studies, >900 participants total8 |
| Pivotal design | Randomized, double-blind, placebo-controlled, multicenter; oral dosing8 |
| Early signal (12 wk) | ~1.8–2.6 kg greater loss vs placebo; 1 mg arm best (METAOD005)8 |
| Primary endpoint (24 wk) | Difference from placebo did NOT reach statistical significance8 |
| With diet + exercise | No detectable added benefit over lifestyle alone8 |
| Outcome | Obesity development halted in 20078 |
| Endocrine profile | No reported rise in IGF-1; no impairment of glucose tolerance13 |
| Cognitive/neurological endpoints | None measured in any trial |
Two honest conclusions follow. First, the compound’s best-evidenced effect — fat loss in obesity — was itself not robustly demonstrated in humans; the trials are more accurately described as showing acceptable short-term safety with disappointing efficacy. Second, and central to this article, none of these studies measured a single neurological or cognitive endpoint. There is no DXA-derived body-composition-versus-cognition analysis, no measure of circulating inflammatory markers linked to cognitive testing, no neuroimaging, and no follow-up of cognitive trajectories. The entire evidence base concerns adipose tissue and body weight in metabolically healthy-to-obese adults. Extrapolating from “did not reliably reduce fat” to “protects the brain” is not a small step; it crosses two disciplines with no data on the far side.
How AOD-9604 Compares With Interventions Studied for the Obese Brain
One of the clearest ways to locate AOD-9604 is to place it beside interventions and compounds that have genuinely been investigated in the context of obesity and brain health. The contrast is instructive not because AOD-9604 competes with them — it has never entered the arena — but because it shows what an actual candidate looks like in terms of mechanism, brain access, and evidence.
| Intervention / class | Mechanism relevant to the obese brain | Level of brain/cognition evidence |
|---|---|---|
| Weight loss via lifestyle / bariatric surgery | Reduces adiposity, systemic inflammation, insulin resistance | Human studies link sustained weight loss to improved cognitive markers; strongest indirect route11 |
| Incretin agonists (e.g., tirzepatide, retatrutide) | Large weight loss; GLP-1 receptors present in brain; possible anti-inflammatory signaling | Robust weight-loss RCTs; neuroprotection an active but unproven research question |
| TLR4 inhibition (e.g., experimental TAK-242) | Blocks microglial inflammatory trigger from fatty acids/LPS | Preclinical (rodent) attenuation of diet-induced neural effects10 |
| Anti-inflammatory / microglial-targeted approaches | Dampen central neuroinflammation directly | Mechanistic and preclinical; not established therapies12 |
| Full-length growth hormone / IGF-1 | Trophic, IGF-1-mediated neuronal support | Studied in some contexts; central effects and risk-benefit uncertain |
| AOD-9604 | Peripheral lipolysis (fat mobilization); IGF-1-sparing by design; no established BBB penetration | No cognitive or neuroprotection studies of any kind |
The pattern is unmistakable. Every serious approach to the obese brain acts on a mechanism plausibly linked to central injury: it reduces adiposity and systemic inflammation durably (lifestyle, surgery, potent incretin agents), blocks the microglial inflammatory trigger, dampens neuroinflammation directly, or supplies trophic support through IGF-1. AOD-9604 does none of these. Its mechanism is directed at peripheral fat cells, it was intentionally uncoupled from IGF-1, it has no established brain access, and it has never been tested for a neurological endpoint.
There is a further lesson embedded in this comparison. Even interventions with unambiguous metabolic efficacy and biological plausibility have not yet earned the label “proven neuroprotectant” in obesity — the translation from metabolic improvement to demonstrated cognitive protection remains difficult and largely unsettled. If purpose-relevant agents with real efficacy still face an open question, a peptide whose signature effect is peripheral fat mobilization, which could not reliably beat placebo even at its intended metabolic job, and which cannot readily reach neurons, has at best a remote and entirely untested claim to relevance here.
None of this makes AOD-9604 uninteresting. As a selective lipolytic probe with an IGF-1-sparing profile, it remains a legitimate research tool for dissecting fat-tissue biology and adrenergic control of lipolysis. It simply means its place is in the study of adipose metabolism, not brain protection. Researchers wanting precise definitions of the terms used here — lipolysis, IGF-1, neuroinflammation, blood-brain barrier — can consult the site’s peptide and metabolic glossary.
Research Models and Methodology
Understanding how AOD-9604 has actually been studied clarifies both what the data can support and what it cannot. The methodology falls into three tiers, none of which addresses the brain.
In vitro and ex vivo fat-tissue work. The foundational metabolic studies examined lipolysis and lipogenesis in adipose tissue and isolated fat cells, measuring free-fatty-acid release, fat oxidation, and expression of enzymes and receptors involved in fat handling.1 These assays are well suited to characterizing a lipolytic agent but say nothing about neurons. A rigorous neuroprotection investigation would instead use neuronal or mixed neuron-glia cultures (for example, primary hippocampal neurons, SH-SY5Y cells, or microglial lines such as BV-2) exposed to relevant insults — palmitate or other saturated fatty acids, lipopolysaccharide, amyloid-β, oxidative or excitotoxic stress — with endpoints such as cell viability, apoptotic markers, reactive-oxygen-species levels, and inflammatory cytokine release. No such experiments with AOD-9604 have been reported.
Rodent models. The most methodologically informative animal work is the β3-AR knockout study, a clean genetic approach that isolated the receptor’s contribution to the compound’s chronic metabolic effects by comparing knockout and wild-type mice under identical treatment.2 Obese-rodent and Zucker fatty-rat models were also used to assess body-weight and lipolytic responses.3 Critically, these are metabolic and obesity models with metabolic endpoints, not neuroscience models. The gold-standard preclinical approach for the obese-brain question uses diet-induced-obesity mice with cognitive testing (Morris water maze, novel-object recognition), quantification of hippocampal microglial activation and cytokines, long-term-potentiation electrophysiology, and BBB-integrity assays — or, for neurodegeneration more broadly, transgenic Alzheimer’s models or ischemia models. AOD-9604 has not, to the available literature, been run through any of these, and no study has measured whether it even reaches brain tissue.
Human trials. The clinical methodology was appropriate for an obesity drug: randomized, double-blind, placebo-controlled, multicenter designs with weight and body-composition endpoints, plus dedicated safety and pharmacokinetic studies.48 A safety and tolerability study specifically characterized the peptide’s behavior in humans.4 But the endpoints were metabolic. None of the human studies enrolled participants for a cognitive question, administered neuropsychological testing, performed neuroimaging, or measured central inflammatory markers.
The methodological bottom line is that AOD-9604’s evidence architecture was built to answer a fat question and was, at that, inconclusive. To answer the neuroprotection question honestly would require an entirely new program: neuronal and microglial insult assays, validated diet-induced-obesity cognitive models with brain-specific and functional endpoints, pharmacokinetic work establishing whether the peptide reaches the CNS at all, and eventually controlled human trials with cognitive outcomes. Until that work exists, any statement about AOD-9604 and neuroprotection is hypothesis, not finding.
Safety and Tolerability
If there is one area where AOD-9604’s clinical record is relatively reassuring, it is short-term safety — a point that must be stated carefully, because “did not appear harmful in obesity trials” is not the same as “safe for use in older or cognitively vulnerable patients,” and short-term tolerability in one population does not license claims of safety in another, let alone claims of benefit.
In the human obesity program, AOD-9604 was generally reported to be well tolerated over the studied durations, with a safety profile that did not raise the endocrine concerns associated with full-length growth hormone. Specifically, the compound was reported not to elevate IGF-1 and not to impair glucose tolerance or insulin sensitivity in the studied settings — the very effects it was engineered to avoid.13 A dedicated human safety and tolerability evaluation supported an acceptable short-term profile at the doses tested,4 and a later characterization framed the ingredient as having a favorable safety and metabolism profile in the sponsor’s view.6
Several important caveats temper this picture, and they bear directly on any imagined neurological use:
- Population mismatch. Safety was established in metabolically healthy-to-obese adults, not in the elderly, people with neurodegenerative disease, or those with cerebrovascular disease — populations with altered pharmacokinetics, polypharmacy, and BBB changes where a peptide’s behavior may differ.
- Duration. Trials ran on the order of weeks to a few months. Neuroprotection is inherently a long-horizon question; the long-term safety of AOD-9604 has not been characterized.
- Route and formulation. The pivotal human work used oral dosing, whereas research use of reconstituted material is typically subcutaneous. Safety data do not transfer automatically across routes.
- Product quality. Much material circulating outside regulated channels is sold as “research chemical” of variable purity. Impurities, endotoxin, and mislabeling are real risks unrelated to the molecule’s intrinsic safety and entirely dependent on sourcing.
- Immunogenicity and characterization concerns. Regulatory review of AOD-9604 for compounding cited inadequate physicochemical characterization and immunogenicity concerns — issues relevant to repeated administration.7
- Sport prohibition. AOD-9604 is prohibited in sport by the World Anti-Doping Agency; for athletes this is a regulatory hazard regardless of pharmacology.9
The reasonable reading is that AOD-9604 has not thrown up major short-term safety signals in the limited, non-neurological populations studied, and that its IGF-1-sparing profile avoids some GH-related concerns. But a clean short-term profile in obese adults provides no assurance about long-term administration to cognitively vulnerable patients, and it certainly provides no evidence of benefit. Absence of demonstrated harm and absence of demonstrated efficacy can coexist, and here they do.
Handling and Reconstitution in a Research Context
Because AOD-9604 is most often encountered as a lyophilized (freeze-dried) powder in a sealed vial, a brief, strictly educational note on laboratory handling is warranted — with the emphasis that this is standard research-peptide practice, not a usage recommendation, and that AOD-9604 is not an approved therapeutic for any indication, neurological or otherwise.
Lyophilized peptides are generally reconstituted with sterile or bacteriostatic water for laboratory purposes. The diluent is directed slowly against the inside wall of the vial rather than sprayed onto the powder, and the vial is gently swirled rather than shaken, because vigorous agitation can shear peptide bonds and denature the material. The volume of diluent chosen simply sets the concentration: a fixed mass of peptide dissolved in a larger volume yields a lower concentration per unit volume, which is the arithmetic underlying any reconstitution chart. General principles of reconstitution and concentration math are described in the site’s peptide reconstitution guide, which is illustrative of how these calculations are typically presented for research compounds.
Stability and storage considerations that recur across the research-peptide literature include:
| Parameter | Typical research-context practice |
|---|---|
| Lyophilized storage | Cool, dark conditions; long-term stability favored by freezing |
| After reconstitution | Refrigerated; used within a limited window |
| Light and heat | Minimize exposure; both can degrade peptides |
| Agitation | Swirl gently; avoid shaking or foaming |
| Freeze-thaw | Repeated cycles degrade peptides; avoid |
| Sterility | Aseptic technique; bacteriostatic water for multi-use practice |
It bears repeating that meticulous handling changes nothing about the evidence question. A perfectly reconstituted, high-purity vial of AOD-9604 is still a compound with zero neuroprotection data. Good technique preserves whatever biological activity the molecule has; it does not create efficacy where none has been demonstrated, and it certainly does not confer brain access the molecule was never shown to possess.
Regulatory Status
AOD-9604’s regulatory picture is layered and frequently misrepresented, so precision matters — especially because regulatory language is often mistaken for evidence of clinical benefit.
No therapeutic approval, anywhere. AOD-9604 is not approved as a drug for obesity, cognitive decline, neurodegeneration, osteoarthritis, or any other condition by the U.S. Food and Drug Administration, the European Medicines Agency, or any comparable major regulator. Its pharmaceutical development for obesity was abandoned in 2007 after the pivotal trial failed to demonstrate a significant benefit over placebo.8 There is, correspondingly, no approved indication of any kind — and certainly none in neurology.
Supplement and food-ingredient framing. After the drug program ended, the compound was repositioned by its sponsor as a metabolic-health ingredient, supported by a self-affirmed “generally recognized as safe” (GRAS) characterization and safety/metabolism publications.6 It is essential to understand that a GRAS self-affirmation addresses ingredient safety at supplement-level exposure; it is emphatically not a finding of efficacy and not drug approval. A GRAS characterization tells you nothing about whether AOD-9604 does anything useful for fat, and less than nothing about the brain — these are unrelated regulatory questions. Conflating “recognized as safe as a food ingredient” with “shown to work as a medicine” is one of the most common errors in the marketing literature around this compound.
U.S. compounding review. In 2024, AOD-9604 was among peptide substances considered by the FDA’s Pharmacy Compounding Advisory Committee for potential inclusion on the Section 503A bulk drug substances list. On December 4, 2024, the committee reviewed both AOD-9604 (free base) and AOD-9604 acetate, and the FDA’s own briefing materials raised concerns about physicochemical characterization, immunogenicity and impurities, and a lack of clinical effectiveness data — a clear signal of continued regulatory caution about compounded peptide products.7 This underscores that, in the United States, AOD-9604 does not occupy a settled, sanctioned place even within the compounding framework.
Anti-doping prohibition. The World Anti-Doping Agency has stated that AOD-9604 is prohibited in sport, falling under the categories covering growth factors and related substances. Athletes subject to WADA-compliant testing should assume that use will constitute an anti-doping rule violation.9
The regulatory synthesis is straightforward: AOD-9604 occupies an ambiguous middle ground — not an approved drug, variously handled as a supplement ingredient, unsettled in U.S. compounding, and banned in sport — with no regulatory recognition of any therapeutic use, and nothing remotely approaching a neurological indication. For any legitimate exploration of the compound in the brain, the appropriate path is formal preclinical and clinical investigation under regulatory oversight, not off-label or informal use. The same standard applies to related growth-factor peptides investigated for cognitive endpoints; for a compound where the age-related cognitive question is being examined more directly, see the analysis of whether sermorelin supports cognitive function in age-related neurodegeneration, which likewise finds the evidence preliminary rather than settled.
Frequently Asked Questions
Does AOD-9604 protect the brain or improve cognition in obesity?
There is no direct evidence that it does. AOD-9604 is a peripheral, fat-cell-targeting fragment of growth hormone that was specifically engineered not to raise IGF-1, one of the main pathways through which growth hormone supports neurons.1 No human cognitive trials, no controlled animal neurodegeneration models, and no neuronal cell studies have tested it for neuroprotection. Claims that it protects the obese brain generally borrow the reputation of full-length growth hormone or of weight loss in general, neither of which AOD-9604 reliably delivers.
Can AOD-9604 even reach the brain?
There is no evidence that it does so in meaningful amounts. Peptides generally do not cross the blood-brain barrier without a specific transport mechanism, and AOD-9604 was characterized as acting peripherally on fat tissue rather than centrally — part of its appeal as an obesity candidate was that it did not depend on central appetite suppression. Any neuroprotective effect would therefore have to be indirect, mediated through peripheral metabolic changes rather than direct action on neurons.
Could reducing body fat with AOD-9604 indirectly protect the brain?
In principle, sustained weight loss and metabolic improvement are associated with better cognitive outcomes in obese populations, so an indirect route is conceivable.11 But that route depends on the compound actually producing durable fat loss, and in its pivotal 24-week human trial AOD-9604 did not separate significantly from placebo on weight.8 The indirect neuroprotection hypothesis fails at its first link because the underlying metabolic effect was not robustly demonstrated.
How does obesity actually damage the brain?
The best-supported model is that obesity produces chronic systemic inflammation, elevated free fatty acids, insulin resistance, and vascular dysfunction, which are transmitted into the brain and drive neuroinflammation — microglial activation and cytokine release, especially in the hypothalamus and hippocampus — impairing synaptic function and memory.1012 Notably, mobilizing fat into circulating free fatty acids is part of the injurious cascade, not obviously part of a solution, which is one reason a purely lipolytic agent is a poor mechanistic fit.
Did AOD-9604 work for weight loss in humans?
Not convincingly. Across roughly six trials in more than 900 participants — a tally from the sponsor’s development-program summary rather than a single publication — an early 12-week study (METAOD005) suggested modestly greater loss than placebo, with a low dose performing best. But the pivotal 24-week study (METAOD006), a randomized, double-blind, placebo-controlled, multicenter trial, did not reach statistical significance at its primary endpoint, and the early signal disappeared against an intensive diet-and-exercise background. Obesity development was halted in 2007.8
Why do some sources imply a growth-hormone peptide should help the obese brain?
That reasoning stitches together two true facts — growth hormone has central effects, and obesity harms the brain — with a false bridge. Growth hormone’s neuronal support flows largely through IGF-1, but AOD-9604 was designed to isolate only the fat-mobilizing tail and was repeatedly reported not to elevate IGF-1.13 Removing the IGF-1 signal removes the principal trophic route, and the compound cannot reliably reach the brain to act by another one.
What is the strongest evidence for AOD-9604 doing anything at all?
Its best-supported mechanistic finding is that its chronic metabolic effects depend on the β3-adrenergic receptor on fat cells, shown by a knockout-mouse study.2 Its clearest regenerative signal is in cartilage, not brain: intra-articular injection improved cartilage scores in a rabbit osteoarthritis model.5 Neither result supports a neuroprotective use.
Could AOD-9604 ever become a neuroprotective therapy?
It cannot be ruled out, but it would require entirely new research — pharmacokinetic work establishing brain access, neuronal and microglial insult assays, validated diet-induced-obesity cognitive models with brain and functional endpoints, and controlled human trials — and it would have to overcome a mechanism directed at fat rather than neurons and a compound that could not reliably beat placebo at its intended metabolic job. Realistically, starting from no data and an unfavorable mechanism, it is a long shot.
Is AOD-9604 approved or legal?
It is not approved as a drug for any condition by the FDA, EMA, or other major regulators. It has been handled as a supplement/food-type ingredient via a self-affirmed GRAS characterization (a safety framing, not efficacy or drug approval), was reviewed with regulatory concern for the FDA’s 503A compounding bulks list in 2024, and is prohibited in sport by WADA.679
References
- Ng FM, Sun J, Sharma L, et al. Metabolic studies of a synthetic lipolytic domain (AOD9604) of human growth hormone. Horm Res. 2000;53(6):274-278. PMID: 11146367. https://pubmed.ncbi.nlm.nih.gov/11146367/
- Heffernan MA, Thorburn AW, Fam B, et al. The effects of human GH and its lipolytic fragment (AOD9604) on lipid metabolism following chronic treatment in obese mice and beta(3)-AR knock-out mice. Endocrinology. 2001;142(12):5182-5189. PMID: 11713213. https://pubmed.ncbi.nlm.nih.gov/11713213/
- Ng FM, Jiang WJ, Gianello R, et al. Molecular and cellular actions of a structural domain of human growth hormone (AOD9401) on lipid metabolism in Zucker fatty rats. J Mol Endocrinol. 2000;25(3):287-298. PMID: 11116208. https://pubmed.ncbi.nlm.nih.gov/11116208/
- Stier H, Vos E, Kenley D. Safety and Tolerability of the Hexadecapeptide AOD9604 in Humans. Journal of Endocrinology and Metabolism. 2013;3(1-2):7-15. https://www.jofem.org/index.php/jofem/article/view/157
- Kwon DR, Park GY. Effect of Intra-articular Injection of AOD9604 with or without Hyaluronic Acid in Rabbit Osteoarthritis Model. Ann Clin Lab Sci. 2015;45(4):426-433. PMID: 26275694. https://pubmed.ncbi.nlm.nih.gov/26275694/
- Moré MI, Kenley D. Safety and Metabolism of AOD9604, a Novel Nutraceutical Ingredient for Improved Metabolic Health. Journal of Endocrinology and Metabolism. 2014;4(3):64-77. https://www.jofem.org/index.php/jofem/article/view/213
- U.S. Food and Drug Administration. Pharmacy Compounding Advisory Committee (PCAC) Briefing Document, December 4, 2024 (AOD-9604 free base and acetate; review for Section 503A bulk drug substances). https://www.fda.gov/media/183584/download
- The effect of AOD9604 on weight loss in obese adults: randomized, double-blind, placebo-controlled, multicenter obesity program (METAOD005/METAOD006; development halted 2007). https://www.researchgate.net/publication/295313034
- World Anti-Doping Agency. WADA statement on the substance AOD-9604. https://www.wada-ama.org/en/news/wada-statement-substance-aod-9604
- Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav Immun. 2014;42:10-21. PMID: 24727365. https://doi.org/10.1016/j.bbi.2014.04.001
- Nguyen JCD, Killcross AS, Jenkins TA. Obesity and cognitive decline: role of inflammation and vascular changes. Front Neurosci. 2014;8:375. PMCID: PMC4237034. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4237034/
- Microglial FABP4-UCP2 Axis Modulates Neuroinflammation and Cognitive Decline in Obese Mice. Int J Mol Sci. 2022. PMCID: PMC9032181. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9032181/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. AOD-9604 is not approved by the FDA, EMA, or any comparable regulator for the treatment, cure, or prevention of cognitive decline, neurodegeneration, obesity, or any other disease, and no neuroprotective or cognitive benefit in obese phenotypes or any other population has been demonstrated in controlled research. Nothing here is medical advice or a recommendation for human use. AOD-9604 is prohibited in sport by WADA. Any legitimate investigation of this compound should occur within properly authorized preclinical or clinical research under appropriate oversight. Readers should consult qualified professionals and applicable regulations before making any decisions.