The question in the title contains a chain of assumptions that deserves to be pulled apart before we accept it. It asks how Selank influences GABA signaling to lower anxiety — which quietly presumes three things: that Selank meaningfully engages the GABA system, that this engagement is the route by which it acts on anxiety, and that its anxiety-lowering effect is itself well established. Each of these is, at best, partially supported and, in one important case, directly contradicted by a peer-reviewed experiment. So rather than narrate a tidy mechanism as though it were settled fact, this article treats the GABA hypothesis as what it actually is: a plausible, frequently repeated, but under-proven story built largely on Russian preclinical work and a small number of open-label or comparator-only human studies.
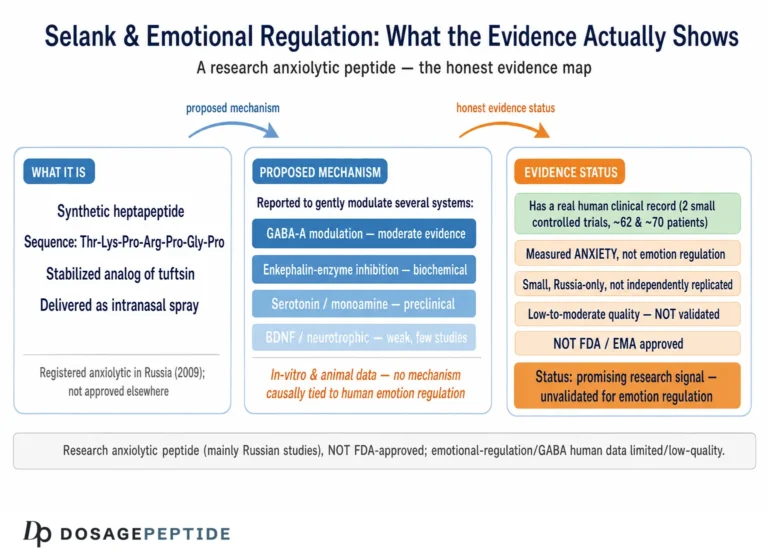
Selank is a synthetic heptapeptide developed in Russia as an anxiolytic and nootropic agent. It is not approved by the U.S. Food and Drug Administration or the European Medicines Agency for anxiety or for anything else; its regulatory home is the Russian pharmacopoeia, and even there the supporting clinical literature is thin by Western standards.1 Most of what is claimed about its GABAergic action traces to a handful of gene-expression studies, one radioligand-binding observation, and an animal experiment pairing it with diazepam — augmented, unhelpfully, by a large volume of commercial copy that states the mechanism with a confidence the primary data do not earn.
This piece is written for researchers and scientifically literate readers who want an honest map: what the GABA hypothesis actually rests on, where the evidence is genuinely suggestive, where it is absent, and where it points the other way. We will cover the compound’s structure and origin, a primer on GABA signaling itself, the specific allosteric-modulation claim, the gene-expression data behind it, a cautionary cell-culture result that found no direct GABA-A effect at all, the diazepam-synergy experiment, the several non-GABA mechanisms that may matter as much or more, the human clinical record and its limits, a comparison with benzodiazepines, laboratory handling, and regulatory status. The guiding principle throughout is restraint. Selank is an investigational peptide with an interesting but immature evidence base, and nothing here should be read as suggesting it is a proven treatment for any anxiety disorder.
What Selank Is and Where It Came From
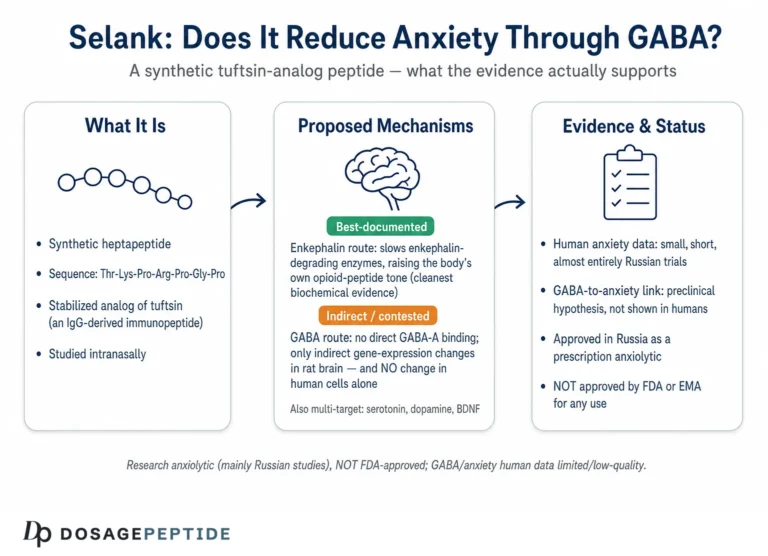
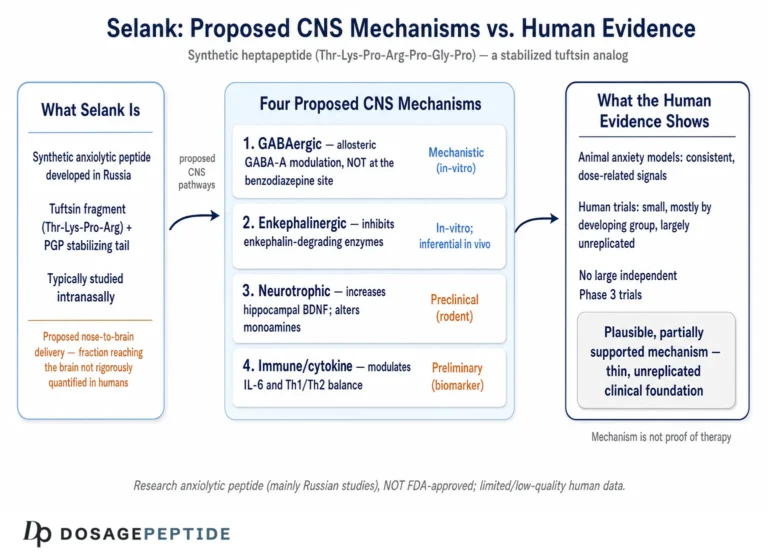
Selank is a synthetic heptapeptide with the amino-acid sequence Thr-Lys-Pro-Arg-Pro-Gly-Pro (TKPRPGP). Its design is a deliberate act of molecular engineering on a natural template. The first four residues — Thr-Lys-Pro-Arg — are the sequence of tuftsin, a naturally occurring tetrapeptide first described in the 1970s that is released from the Fc portion of immunoglobulin heavy chains and that carries immunomodulatory activity, stimulating macrophage phagocytosis and influencing cytokine balance.2 Tuftsin by itself is a poor drug candidate for the brain: it is rapidly degraded by peptidases and has a fleeting biological life. The Russian designers appended a C-terminal tripeptide tail, Pro-Gly-Pro, to the tuftsin core. That addition is the whole trick. Proline-rich termini resist enzymatic cleavage, so the Pro-Gly-Pro tail acts as a protective cap that dramatically extends the molecule’s stability and duration of action while, as it turns out, also shifting its pharmacology away from tuftsin’s purely immune profile toward the central-nervous-system effects that made Selank interesting as a neuropsychiatric candidate.1
The compound was developed at the Institute of Molecular Genetics of the Russian Academy of Sciences, the same research milieu that produced the related peptide Semax (a fragment of adrenocorticotropic hormone). Both belong to a Russian research program built around the idea of turning short, naturally derived regulatory peptides into stabilized drug-like molecules — what its own authors have described as “a new generation of drugs” based on natural regulatory peptides.1 Understanding this lineage matters, because it explains a recurring feature of the Selank literature: the great majority of it originates from a small number of affiliated Russian groups, is often published in Russian-language journals, and has seen little to no independent replication in Western laboratories. That is not, by itself, evidence that the findings are wrong. But it is a structural weakness in the evidence base that any honest assessment has to keep in view.
Pharmacologically, Selank is usually administered in research and clinical settings intranasally, a route that bypasses gastric peptidases and offers partial direct nose-to-brain transport alongside systemic absorption across the nasal mucosa. One of the puzzles of the compound is the mismatch between its very short measured plasma presence — the intact peptide is cleared within minutes — and behavioral and biochemical effects that persist for hours. The leading explanation is that Selank is not simply cleared but processed: peptidases progressively trim the Pro-Gly-Pro tail to release shorter fragments, some tuftsin-like, that retain biological activity, so that the administered heptapeptide functions partly as a pro-drug for a family of active metabolites.1 This detail becomes relevant later, because it complicates any clean statement about “what Selank does at the receptor” — the molecule that reaches a given synapse may not be the molecule that was administered.
There is one further consequence of the tuftsin heritage that the GABA-centric framing tends to bury. Because its core sequence is tuftsin, Selank retains immunomodulatory properties — it has been reported to influence the expression of interleukin-6 and to shift the balance of T-helper-cell cytokines.1 This matters more than it might first appear, because the relationship between systemic inflammation and anxiety is now well recognized: pro-inflammatory cytokines can act on the brain to worsen anxious and depressive states, and agents that dampen that signaling can, in principle, exert an indirect anxiolytic effect. If part of what Selank does is modulate neuroimmune signaling, then attributing its anxiolysis wholly to GABA is not just incomplete but potentially misleading — the calming effect might arise partly upstream of any neurotransmitter system, in the cytokine milieu that sets the brain’s inflammatory tone. This is speculative, but it is grounded in the compound’s documented immune activity, and it is one more reason to resist a single-mechanism story.
For readers who want the broader neurobehavioral picture of the compound rather than the narrow GABA question examined here, the companion article on how Selank regulates behavior via central nervous system pathways surveys its wider CNS profile, and general terminology is defined in the site’s peptide glossary. Here the focus is deliberately tight: the specific claim that Selank works through GABA.
A Primer on GABA Signaling and Why It Matters for Anxiety
To evaluate whether Selank acts through GABA, it helps to be precise about what that would actually mean. Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter of the mammalian central nervous system. Where glutamate excites, GABA restrains; the balance between the two sets the overall tone of neural circuits, and anxiety states are strongly associated with a shift toward under-inhibition — too little GABAergic braking on circuits that generate fear and arousal, particularly in the amygdala and its cortical connections.3
GABA acts through two receptor families that must not be conflated. GABA-B receptors are metabotropic, G-protein-coupled receptors that produce slower, modulatory effects. GABA-A receptors, which are the ones relevant to the Selank anxiolytic story, are ionotropic: they are ligand-gated chloride channels assembled from five subunits drawn from a large family (α1–6, β1–3, γ1–3, and others), most commonly in a 2α:2β:1γ arrangement.3 When GABA binds at the interface between α and β subunits, the channel opens and chloride ions flow according to their electrochemical gradient, typically hyperpolarizing the neuron and making it harder to fire. That is the molecular substance of “inhibition.”
The reason this matters for anxiety is that the most established anxiolytic drugs in medicine — the benzodiazepines — work precisely here. A benzodiazepine such as diazepam does not open the channel itself; it binds at a distinct allosteric site located at the interface between an α and the γ subunit and increases the receptor’s response to GABA, enhancing the frequency of channel opening when GABA is present.3 This is the definition of a positive allosteric modulator: it does not activate the receptor directly but potentiates the endogenous ligand’s effect. The clinical properties of benzodiazepines — anxiolysis, sedation, muscle relaxation, anticonvulsant activity, and, unfortunately, tolerance and dependence — all flow from this potentiation, with different subunit compositions (notably which α subtype is present) governing which effects predominate.3
One more layer of GABA biology is worth spelling out, because it becomes decisive later. GABA-A receptors do not all behave the same way, and the difference is governed largely by which subunits they contain. Synaptic receptors, typically containing a γ2 subunit, mediate fast, transient “phasic” inhibition in response to bursts of released GABA. A separate population of extrasynaptic receptors, often containing a δ subunit, mediates slow, sustained “tonic” inhibition driven by low ambient GABA concentrations, and these tonic receptors are strongly implicated in the regulation of anxiety and vigilance.3 Because subunit composition dictates both the pharmacology and the physiology of a given receptor, a modulator can be exquisitely selective — potent at receptors carrying one subunit combination and inert at another. That principle is not an academic aside here; it is precisely the explanation the Russian investigators would later invoke to reconcile a cell-culture experiment that found no Selank effect with brain data that suggested one.
This background frames the Selank hypothesis sharply. To claim that Selank “influences GABA signaling to lower anxiety” in a benzodiazepine-like way is to make a specific, testable assertion: that the peptide changes how GABA-A receptors respond to GABA, presumably by an allosteric mechanism distinct from the classical benzodiazepine site (since Selank conspicuously lacks the sedative and dependence liabilities that define benzodiazepine action). The interesting question is not whether such a mechanism is conceivable — it plainly is — but whether the actual data demonstrate it, merely suggest it, or fail to find it. As we will see, the literature contains all three.
The Central Hypothesis: Allosteric Modulation of GABA-A Affinity
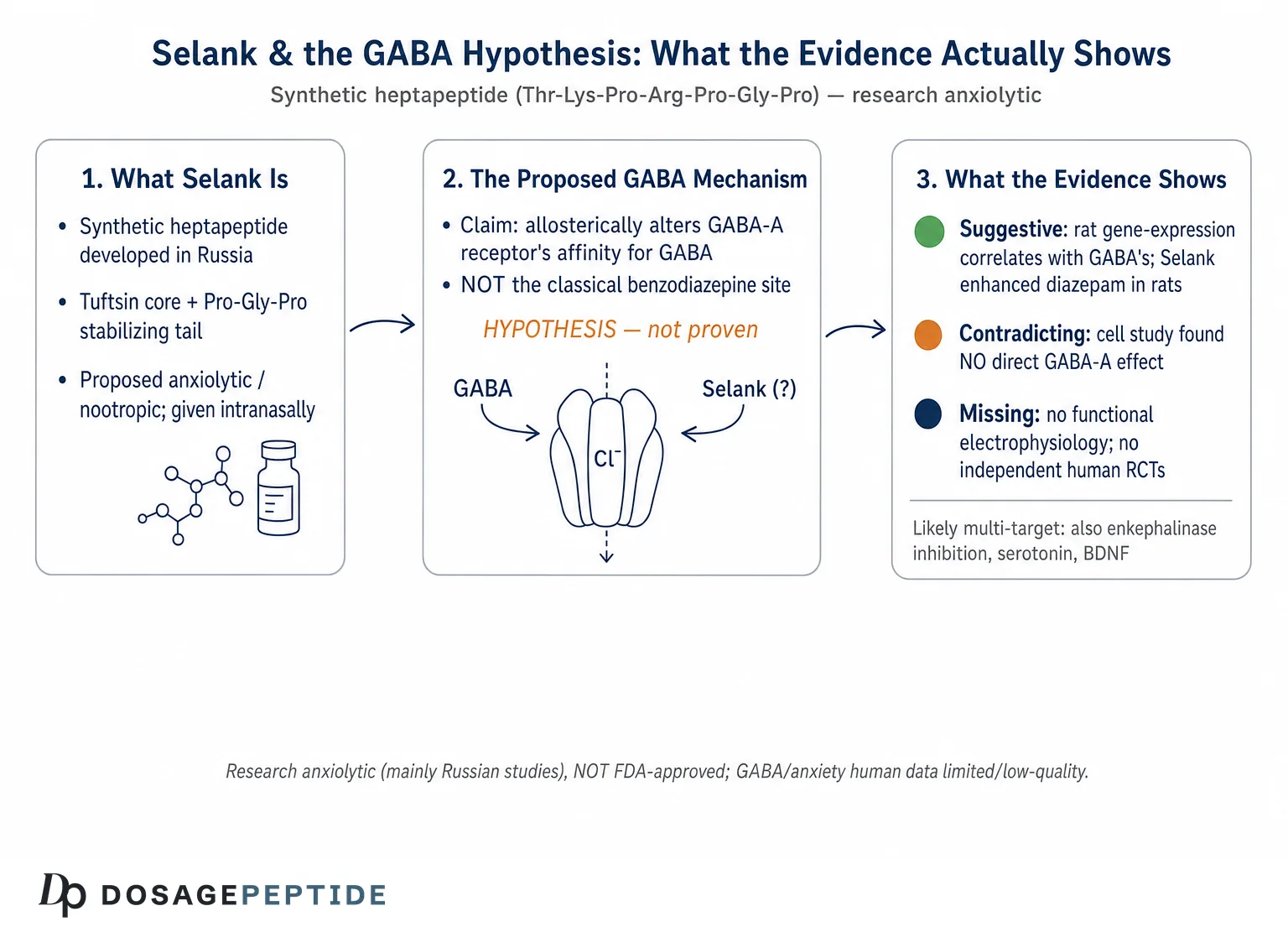
The specific GABA claim made for Selank is not that it binds where GABA binds, nor that it binds the benzodiazepine site. It is subtler: that Selank allosterically alters the affinity of the GABA-A receptor for GABA itself. In the words of the Russian gene-expression work, Selank appears to act “not directly through the center of the specific binding of GABA, but rather allosterically by altering the affinity of the GABA receptor for GABA.”4 A parallel formulation from the diazepam-synergy study is that Selank “affects the specific binding of GABA to GABA-A receptors,” possibly by changing the affinity of endogenous ligand for the receptor.5
It is worth being clear about what kind of claim this is. A change in receptor affinity for an endogenous transmitter is a legitimate and well-precedented mode of allosteric action; many modulators work this way. But affinity is a biophysical parameter that is most convincingly demonstrated by direct radioligand-binding assays across a range of ligand concentrations, ideally with functional electrophysiology (measuring chloride current in response to GABA with and without the modulator) to confirm that the binding change translates into a change in channel behavior. The Selank literature does not, to the available published record, contain that gold-standard functional demonstration. What it contains instead is a binding observation and, more extensively, transcriptional evidence — changes in the expression of GABA-system genes — from which the affinity hypothesis is inferred.45
That distinction — between a demonstrated functional modulation of the receptor and an inferred one built largely on downstream gene-expression changes — is the single most important thing to hold onto in this whole discussion. A compound that changes the expression of GABA-receptor subunit genes is doing something to the GABA system, but that is not the same as, and does not by itself establish, direct allosteric potentiation of the channel in the way a benzodiazepine potentiates it. The popular shorthand that “Selank is a positive allosteric modulator of GABA-A at a non-benzodiazepine site” — a phrasing that circulates widely in commercial and secondary sources, sometimes attached to a precise-sounding but unsourced figure such as a “38% increase in GABA-A binding” — overstates what the primary literature actually shows. The honest version is that Selank plausibly modulates GABA-A signaling by an indirect, possibly expression-linked mechanism, and that the exact biophysical details remain unresolved.
Reading the Gene-Expression Evidence
The most substantial pillar under the GABA hypothesis is a body of transcriptional work examining how Selank changes the expression of neurotransmission-related genes in rat brain. The clearest single study administered Selank (and, comparatively, GABA) to rats and used real-time PCR to measure the expression of 84 genes involved in neurotransmission — GABA-receptor subunits, transporters, ion channels, and dopamine and serotonin receptors — in the frontal cortex at one and three hours after administration.4
The findings were substantial in scale. At one hour, 45 of the 84 genes showed significant changes in expression; by three hours, 22 genes were altered.4 The result most often cited in support of the GABA hypothesis is a correlation: at the one-hour timepoint, the pattern of expression changes produced by Selank correlated strongly with the pattern produced by GABA itself, with a reported correlation coefficient of about r = 0.86.4 The interpretation offered by the authors is that Selank and GABA act on overlapping molecular machinery — that Selank’s transcriptional signature resembles GABA’s because the peptide is engaging the same system, consistent with an allosteric influence on GABA-A signaling. Specific GABA-related transcripts, including subunit genes such as Gabre and Gabrq, showed pronounced dynamic changes, sharply down at one hour and up by three.4
Taken at face value, this is genuinely suggestive: it is difficult to explain a strong correlation between the transcriptional effects of a peptide and those of GABA without some functional relationship between them. But several honest caveats temper how much weight it can bear. First, gene expression is a downstream readout, temporally and mechanistically removed from the ion channel; a compound can change subunit-gene transcription through many routes, including secondary responses to altered neuronal activity, without directly modulating the existing receptor. Second, a correlation of expression patterns establishes similarity, not the direction or mechanism of causation. Third, the work is single-laboratory-lineage and has not been independently replicated in Western labs, a recurring limitation across the Selank corpus. The transcriptional data are the best evidence for a GABA connection — and they remain a step or two removed from proving the receptor-level mechanism the title implies.
| Proposed mechanism | What the data show | Evidence level |
|---|---|---|
| Allosteric change in GABA-A affinity for GABA | Inferred from a radioligand-binding observation and expression data; no published functional electrophysiology45 | Weak–moderate, indirect |
| Change in GABAergic gene expression (rat cortex) | 45 genes altered at 1 h, 22 at 3 h; r ≈ 0.86 correlation with GABA’s own signature4 | Moderate but single-lineage, unreplicated |
| Direct effect on GABA-A receptors in neuronal cells | None detected in IMR-32 cells; no change in studied gene mRNA from Selank alone6 | Negative result |
| Potentiation of a benzodiazepine (diazepam) | Combined Selank+diazepam reduced anxiety more than either alone in stressed rats5 | Suggestive, animal only |
| Inhibition of enkephalin-degrading enzymes | Dose-dependent inhibition in human serum, IC50 ≈ 15 µM7 | Biochemical, in vitro |
| Serotonergic modulation | Enhanced brain-stem 5-HT metabolism in PCPA-treated rats8 | Preclinical |
| BDNF upregulation (hippocampus) | Increased Bdnf mRNA at 3 h and BDNF protein at 24 h after intranasal dosing9 | Preclinical |
A Cautionary Counter-Result: the IMR-32 Cell Study
Any honest treatment of the GABA hypothesis has to confront a study that cuts against it. In 2017, a group from the same Russian research lineage examined the effects of GABA, Selank, and the antipsychotic olanzapine on the expression of GABAergic-neurotransmission genes in IMR-32 cells, a human neuroblastoma cell line used as a neuronal model.6 The design was well suited to isolate a direct cellular effect of Selank on the GABA system, because it removed the whole-brain context and looked at what the peptide did to GABA-related gene expression in defined neuronal cells.
The result was striking in its plainness: Selank alone produced no changes in the mRNA levels of the genes studied.6 GABA itself altered fourteen genes; but the peptide, on its own, moved none of them. More pointedly, when Selank was combined with GABA, it did not add to GABA’s effect — it largely suppressed it, bringing the GABA-induced expression changes back toward baseline. The authors’ own conclusion was blunt: Selank had “no direct effect on GABA-A receptors” presented in the IMR-32 culture, which they attributed to the particular subunit composition of the receptors expressed in that cell line.6
How should a careful reader reconcile this with the rat-cortex correlation? Several readings are possible, and honesty requires holding them together rather than choosing the most convenient. One is that Selank’s GABA effect is genuinely dependent on receptor subunit composition: if the peptide modulates only certain subunit combinations, a cell line lacking those subunits would show nothing, while intact cortex — with its full diversity of receptor subtypes — would respond. That reading actually rescues and refines the hypothesis rather than demolishing it, and it is the interpretation the authors themselves lean toward. A second, more skeptical reading is that the whole-brain transcriptional correlation reflects indirect, network-level consequences of Selank acting somewhere other than GABA-A receptors, and that when you strip away the network and test the receptors directly in a dish, the direct effect simply is not there. A third possibility is that both are partly true and the GABA contribution is real but modest and context-dependent.
The methodological lesson of the IMR-32 result is broader than Selank. Negative findings in a defined system are, in one sense, more informative than positive findings in a complex one, because they are harder to explain away: a whole brain offers a thousand indirect routes by which a signal might propagate, whereas a dish of neuroblastoma cells with a known receptor complement does not. When Selank moved nothing on GABA-A-related transcripts in that controlled setting, it told us that whatever the peptide does to the GABA system in an intact animal, it is unlikely to be a straightforward, cell-autonomous action on the receptor of the sort a small-molecule potentiator would produce. The most charitable and probably most accurate reconciliation — subunit-dependence — is testable: it predicts that Selank should show an effect in cells engineered to express the specific α/β/γ or δ combinations found in anxiety-relevant circuits, and no effect in cells lacking them. That experiment does not appear to have been done, and until it is, the subunit-dependence explanation remains a reasonable hypothesis rather than a demonstrated fact. This is the recurring shape of the Selank literature: each promising result is followed not by confirmation but by a plausible caveat and an untested next step.
What is not defensible is to cite the rat correlation as proof of a GABA mechanism while ignoring the cell study that found no direct receptor effect. The two results coexist in the same small literature, produced by overlapping investigators, and the intellectually honest summary is that Selank’s relationship to GABA-A receptors is real enough to keep studying but too unresolved to state as established mechanism — and certainly too unresolved to justify the confident receptor-level claims found in marketing copy.
Selank and Diazepam: the Synergy Experiment
The most direct behavioral test of a functional GABA link comes from an experiment pairing Selank with the benzodiazepine diazepam in rats subjected to unpredictable chronic mild stress, a standard model for inducing anxiety- and depression-like states.5 The logic is elegant: if Selank modulates the same GABA-A system that diazepam potentiates, then combining the two should produce effects that reveal an interaction — either additive or synergistic — and might allow a lower benzodiazepine dose to achieve the same anxiolysis.
That is roughly what the study reported. Under chronic stress, combining Selank with diazepam reduced anxiety-like behavior more effectively than either agent given alone, with the combination bringing measures closer to pre-stress baseline than monotherapy achieved.5 The authors interpreted this as consistent with Selank affecting GABA-A binding in a way that enhances diazepam’s receptor action, and framed the practical implication as a possible benzodiazepine dose-sparing strategy — achieving anxiolysis with less benzodiazepine, and therefore potentially less sedation, tolerance, and withdrawal liability.5
This is the strongest single piece of functional evidence for a GABA link, and it deserves to be taken seriously. But its limits are equally important. It is an animal study, in rodents, using behavioral endpoints rather than direct measurement of receptor function; a behavioral interaction between two anxiolytics is compatible with a shared GABA mechanism but does not prove one, because two compounds acting on different anxiety-relevant systems (say, one on GABA-A and one on enkephalin or serotonin pathways) can also produce greater-than-single-agent effects simply by hitting the problem from two angles. The synergy is real and interesting; whether it demonstrates that Selank works through GABA-A specifically, as opposed to alongside it, is not settled by this experiment alone. As with the rest of the corpus, it is single-lineage and awaits independent replication.
The dose-sparing implication deserves a further word of caution, because it is the part most likely to be over-read. A finding that adding Selank lets a lower dose of diazepam achieve a given anxiolytic effect in stressed rats is genuinely appealing — benzodiazepine dose reduction is a real clinical goal, since the sedation, cognitive blunting, tolerance, and dependence that limit these drugs are all dose-related. But an adjunct effect in a rodent stress model is a long way from a validated human dose-sparing regimen. It does not establish that the interaction holds in people, at clinically relevant doses, over the timescales at which benzodiazepine problems actually emerge; nor does it rule out the possibility that co-administering two centrally active agents introduces its own risks. Reading the experiment as support for a specific human combination protocol would be exactly the kind of overreach this article is trying to guard against. It is a mechanistic clue and a hypothesis generator, not a clinical recommendation.
Is GABA Even the Primary Mechanism? Enkephalins, Serotonin, and BDNF
Here is the part of the story that the title’s framing tends to obscure. Even within the Selank literature itself, GABA is not obviously the leading candidate for the compound’s anxiolytic action. Several other mechanisms are at least as well documented, and one of them — the enkephalin pathway — arguably has cleaner biochemical evidence than the GABA hypothesis does.
Enkephalin-degrading enzymes. One of the earliest and most concrete mechanistic findings is that Selank inhibits the enzymes that break down enkephalins, the endogenous opioid peptides involved in stress, pain, and mood regulation. In human serum, Selank dose-dependently inhibited the enzymatic hydrolysis of enkephalin with an IC50 of about 15 µM, more potently than the reference peptidase inhibitors bacitracin and puromycin.7 The clinical hook is notable: patients with generalized anxiety were found to have shortened enkephalin half-life and reduced enkephalinase-inhibiting capacity in blood, and the authors proposed that Selank’s anxiolytic effect may work by restoring enkephalin tone — slowing the breakdown of these calming endogenous peptides.7 This is a biochemically direct, dose-quantified mechanism, and it has nothing to do with GABA receptors.
Serotonin. Selank also modulates the serotonergic system. In rats depleted of serotonin with para-chlorophenylalanine (PCPA), Selank — unlike its parent tuftsin — enhanced serotonin metabolism in the brain stem, altering the 5-HIAA/5-HT turnover ratio in relevant regions.8 Because serotonergic signaling is central to the action of SSRIs and to anxiety regulation generally, this is a plausible independent contributor to any mood effect, and it is a point of pharmacological divergence from tuftsin that the Pro-Gly-Pro modification apparently introduced.
BDNF and neuroplasticity. A third arm is neurotrophic. A single intranasal dose of Selank increased brain-derived neurotrophic factor (BDNF) messenger RNA in the rat hippocampus within about three hours and raised BDNF protein by 24 hours.9 BDNF supports neuronal plasticity and has been repeatedly linked to the mechanisms of antidepressant and anti-anxiety action. A slower, plasticity-based contribution of this kind would help explain why Selank’s reported behavioral effects can outlast its brief plasma presence — and, again, it operates independently of GABA-A receptor potentiation.
It is worth pausing on why this multiplicity is not merely a hedge but the most likely truth. Short regulatory peptides of this kind rarely act like a synthetic small-molecule drug designed to hit a single target with high affinity. They tend instead to nudge several endogenous systems at once, often at modest potency, precisely because they are derived from molecules the body already uses as signals. Selank’s enzymatic fate reinforces the point: as the Pro-Gly-Pro tail is trimmed to release tuftsin-like fragments, the pharmacological population acting in the brain becomes a mixture rather than a single species, and those fragments can carry their own immune and neural activities.12 A compound that is really a small family of related actives, each touching several systems weakly, is exactly the kind of agent for which a clean “the mechanism is X” sentence will always be wrong. The scientifically honest description is distributed and probabilistic: some contribution from enkephalin preservation, some from serotonergic and neurotrophic effects, some possibly from GABAergic modulation, and some from neuroimmune signaling, with the relative weights unknown and likely varying by dose, route, timing, and the anxiety phenotype being studied.
The upshot is that Selank is, at best, a multi-target compound, and the GABA arm is only one of several — and not the most cleanly demonstrated one. A reader who arrives believing “Selank is a GABA drug” should leave understanding that the enkephalin-enzyme mechanism is at least as well evidenced, that serotonin and BDNF are credible parallel routes, and that the compound’s anxiolytic effect (to whatever extent it is real) is probably the sum of several modest actions rather than a single benzodiazepine-like hit on GABA-A. This polypharmacology is, incidentally, a common theme across regulatory peptides; other multi-system agents such as BPC-157 are documented to touch several pathways at once, and the site’s central dosages index frames these molecules as multi-system rather than single-target agents.
What the Human Trials Actually Show — and Their Limits
If the mechanism is unsettled, what about the clinical bottom line: does Selank lower anxiety in people? The honest answer is that a small human literature suggests it might, that this literature is of low methodological quality by contemporary standards, and that it has not been reproduced outside its country of origin.
The most cited human study is a clinical-biological investigation of Selank in generalized anxiety disorder and neurasthenia. It enrolled 62 patients, of whom 30 received Selank and 32 received the benzodiazepine medazepam as an active comparator, with outcomes assessed on standard instruments including the Hamilton Anxiety Rating Scale, the Zung self-rating scale, and clinical global impression.10 The reported result was that Selank produced an anxiolytic effect broadly comparable to the benzodiazepine comparator, while additionally showing antiasthenic and mild psychostimulant effects — that is, without the sedation associated with benzodiazepines — and the authors linked the response to the enkephalin mechanism described earlier.10
On its face this is an encouraging signal. But its limitations are serious and must be stated plainly. The sample is small (dozens, not hundreds). Crucially, the study compared Selank against an active drug rather than against placebo, so it can speak to relative performance but not to the placebo-subtracted effect size that regulators and evidence-based medicine require — and anxiety disorders are notorious for large placebo responses. The trial was conducted within the developer’s own research ecosystem, raising the ordinary concerns about independence and publication bias that attend a literature dominated by affiliated groups. It was, by modern reporting standards, incompletely described. And there is essentially no large, randomized, double-blind, placebo-controlled, independently conducted trial of Selank for any anxiety indication in the Western peer-reviewed literature. The result is a compound that is approved and used in one country on the strength of an evidence base that would not, as it stands, support approval by the FDA or EMA.
It is also worth being explicit about what a convincing human evidence base for the GABA-anxiety claim would actually look like, because the gap is instructive. It would include multiple randomized, double-blind, placebo-controlled trials, adequately powered, with pre-registered primary endpoints on validated anxiety instruments, conducted by investigators independent of the developer, and ideally replicated across more than one country and research culture. For a mechanistic claim specifically, it would be strengthened by human biomarker or neuroimaging work — for example, evidence that Selank changes GABAergic tone measurable by magnetic resonance spectroscopy, or that its anxiolytic effect is blunted by a GABA-A antagonist. None of that exists for Selank. What exists is a small comparator study, a scattering of Russian reports on cognitive and anxiety endpoints, and a preclinical literature. That is enough to justify interest and further study; it is not enough to justify the confident therapeutic and mechanistic claims that circulate in consumer-facing material. Holding those two judgments together — genuine scientific interest, genuinely insufficient proof — is the whole discipline of reading this literature well.
The fair synthesis is therefore twofold. First, there is a plausible and internally consistent story — small human studies, animal models, and several candidate mechanisms all pointing in the same anxiolytic direction — that makes Selank a legitimate object of continued research. Second, that story falls well short of proof: the human data are limited and comparator-only, the mechanism (including the GABA arm this article set out to examine) is under-characterized, and independent replication is largely absent. Both things are true at once, and it does no one a service to collapse them into either uncritical endorsement or blanket dismissal.
Selank Versus Benzodiazepines: a Careful Comparison
Because the title invokes GABA — the benzodiazepines’ own territory — a side-by-side comparison clarifies both what the two have in common and where the analogy breaks down. The comparison should be read as a map of hypotheses and evidence levels, not as a claim that Selank is an equivalent or interchangeable anxiolytic.
| Feature | Benzodiazepines (e.g., diazepam) | Selank |
|---|---|---|
| GABA-A relationship | Well-defined positive allosteric modulator at the α/γ benzodiazepine site3 | Proposed indirect/allosteric influence on GABA-A affinity; not demonstrated by functional electrophysiology46 |
| Primary evidence type | Extensive human RCTs, decades of clinical use | Small comparator human studies plus Russian preclinical work10 |
| Sedation | Common, dose-dependent | Reported minimal; possible mild stimulant/antiasthenic effect10 |
| Tolerance / dependence | Well documented | Not reported in available studies (but long-term data lacking) |
| Other mechanisms | Essentially GABA-A specific | Enkephalinase inhibition, serotonergic, BDNF — multi-target789 |
| Regulatory status | Approved worldwide as prescription medicines | Approved in Russia; not FDA/EMA approved for any use11 |
Two honest conclusions come out of this table. First, the frequently drawn equation — “Selank is like a benzodiazepine but without the downsides” — is a marketing simplification that runs ahead of the data. It is true that Selank has not shown the sedation and dependence that limit benzodiazepines, and if that holds up it would be genuinely valuable. But the benzodiazepine comparison also implies an efficacy and mechanistic clarity that Selank has not earned: benzodiazepines have an unambiguous, electrophysiologically proven GABA-A mechanism and an enormous human evidence base, and Selank has neither. Second, the very feature that makes Selank attractive — the absence of classical benzodiazepine liabilities — is itself evidence that it is not simply acting at the benzodiazepine site, which points back to the multi-mechanism, indirect picture this article has developed. The lack of sedation is not a free lunch bolted onto a benzodiazepine mechanism; it is a clue that the mechanism is different, and probably distributed across several systems.
Handling and Reconstitution in a Research Context
Because Selank is typically supplied as a lyophilized (freeze-dried) powder for laboratory use, a brief, strictly educational note on handling is warranted — with the emphasis that this describes standard research-peptide practice and is not a recommendation for human use, and that Selank is not an approved therapeutic in most jurisdictions.
Lyophilized peptides are generally reconstituted with sterile or bacteriostatic water for laboratory purposes. The diluent is directed slowly against the inner wall of the vial rather than sprayed onto the powder, and the vial is swirled gently rather than shaken, because vigorous agitation can shear peptide bonds and denature the material. The chosen volume of diluent simply sets the concentration: a fixed mass of peptide dissolved in a larger volume yields a lower concentration per unit volume, the arithmetic that underlies any reconstitution chart. General walkthroughs of this math and technique appear in the site’s peptide reconstitution guide, which is educational rather than prescriptive.
| Parameter | Typical research-context practice |
|---|---|
| Lyophilized storage | Cool, dark conditions; long-term stability favored by freezing |
| After reconstitution | Refrigerated; used within a limited window |
| Light and heat | Minimize exposure; both accelerate peptide degradation |
| Agitation | Swirl gently; avoid shaking or foaming |
| Freeze–thaw | Repeated cycles degrade peptides; avoid |
| Route in studies | Commonly intranasal in Selank research, exploiting nose-to-brain transport |
It bears repeating that meticulous handling changes nothing about the evidence question. A perfectly reconstituted, high-purity vial of Selank is still a compound whose GABA mechanism is unproven and whose human anxiety data are limited. Good technique preserves whatever biological activity the molecule has; it does not create efficacy or resolve mechanistic uncertainty. A further real-world caveat is that much material sold outside regulated channels as “research chemical” varies in purity and provenance, so impurities and mislabeling are practical confounders that have nothing to do with the molecule’s intrinsic pharmacology and everything to do with sourcing.
Regulatory Status and Evidence Quality
Selank’s regulatory picture is easy to misstate, so precision matters. It is approved and marketed in Russia as an anxiolytic; it is not approved by the U.S. FDA, the European Medicines Agency, or comparable major regulators for anxiety or for any other indication.11 Approval in one national system on the basis of that system’s evidence standards is not equivalent to the multi-trial, placebo-controlled evidence that Western regulators require, and the gap between the two is precisely the gap in the human data described above.
In the United States, Selank has also passed through the recent regulatory scrutiny of compounded peptides. It was placed in Category 2 of the FDA’s interim list of bulk drug substances for use in 503A compounding — the category for substances that may present significant safety risks and that therefore may not generally be compounded — before its nomination was subsequently withdrawn, removing it from that specific list without conferring any approval.11 The practical meaning is that Selank does not have a settled, sanctioned place in U.S. therapeutics, and its regulatory trajectory reflects continued official caution about unapproved peptide products rather than any endorsement of efficacy.
Standing back, the evidence quality for the specific proposition in the title — that Selank influences GABA signaling to lower anxiety — is best summarized in tiers. The anxiolytic effect in humans: plausible but supported only by small, comparator-only, largely unreplicated studies. The GABA mechanism: suggested by rat gene-expression correlations and a diazepam-synergy experiment, undercut by a cell study that found no direct GABA-A effect, and never confirmed by functional receptor electrophysiology. The competing mechanisms (enkephalinase inhibition, serotonin, BDNF): at least as well documented and possibly more central. For any legitimate advance, the path is the ordinary one — direct functional studies of Selank on defined GABA-A receptors, and adequately powered, placebo-controlled, independently conducted human trials — not extrapolation from the current, immature base.
Frequently Asked Questions
Does Selank actually work through GABA to reduce anxiety?
The GABA route is a leading hypothesis, not an established fact. Rat gene-expression studies show that Selank’s transcriptional effects on neurotransmission genes correlate strongly with GABA’s own effects, and a rat experiment found that Selank enhanced the anxiolytic action of the benzodiazepine diazepam.45 But a cell-culture study found no direct effect of Selank on GABA-A receptors, and no published functional electrophysiology demonstrates that Selank modulates the chloride channel the way a benzodiazepine does.6 So the honest position is that Selank probably influences GABA signaling indirectly and partially, but the receptor-level mechanism is unproven.
Is Selank a positive allosteric modulator of GABA-A like a benzodiazepine?
Not in the demonstrated sense that benzodiazepines are. The proposal is that Selank allosterically alters the GABA-A receptor’s affinity for GABA rather than binding the classical benzodiazepine site.45 That claim rests on binding and expression data, not on the functional channel recordings that define benzodiazepine modulation. Confident statements online that Selank is a “non-benzodiazepine positive allosteric modulator,” sometimes with a specific percentage figure attached, overstate the primary evidence.
If not GABA, what else might explain Selank’s effects?
Several mechanisms are at least as well evidenced. Selank inhibits enkephalin-degrading enzymes in human serum, which could raise levels of calming endogenous opioid peptides;7 it enhances serotonin metabolism in rat brain stem;8 and it raises hippocampal BDNF, a plasticity factor linked to mood regulation.9 The anxiolytic effect, to the extent it is real, is probably the sum of several modest actions rather than a single GABA hit.
Has Selank been proven to lower anxiety in humans?
Only weakly. The main human study enrolled 62 patients with generalized anxiety and neurasthenia and reported an anxiolytic effect comparable to the benzodiazepine medazepam, without sedation.10 But it was small, compared Selank to an active drug rather than to placebo, and came from the developer’s own research ecosystem. There is no large, independent, placebo-controlled trial, so the human evidence is suggestive but low-quality.
Why is so much of the Selank research from Russia?
Selank was developed at the Institute of Molecular Genetics of the Russian Academy of Sciences, and nearly all of its literature comes from that lineage of affiliated groups, often in Russian-language journals.1 This concentration is a real limitation: findings have seen little independent Western replication, which weakens confidence even where individual results look promising.
Is Selank approved by the FDA?
No. Selank is approved and used in Russia but is not approved by the U.S. FDA or the European Medicines Agency for anxiety or any other condition.11 In the United States it was placed on, then removed (by nomination withdrawal) from, the FDA’s Category 2 interim list for 503A compounding — a process that reflects caution about unapproved peptides, not approval.
Does Selank cause dependence or withdrawal like benzodiazepines?
Available studies have not reported the tolerance, dependence, or sedation that characterize benzodiazepines, which is one of the compound’s most cited attractions.10 However, long-term and independent safety data are lacking, so the absence of reported problems is not the same as demonstrated long-term safety. It also hints that Selank is not simply acting at the benzodiazepine site.
How is Selank handled in a research setting?
As a lyophilized powder, it is reconstituted with sterile or bacteriostatic water using gentle technique (swirl, do not shake), stored cool and dark, protected from freeze–thaw cycles, and in Selank studies is often administered intranasally to exploit nose-to-brain transport.1 Handling quality preserves activity but has no bearing on the open questions about mechanism or human efficacy.
References
- Kolomin T, Shadrina M, Slominsky P, Limborska S, Myasoedov N. A new generation of drugs: synthetic peptides based on natural regulatory peptides. Neuroscience & Medicine. 2013;4(4):223-252. doi:10.4236/nm.2013.44035. https://www.scirp.org/journal/paperinformation?paperid=40708
- Siemion IZ, Kluczyk A. Tuftsin: on the 30-year anniversary of Victor Najjar’s discovery. Peptides. 1999;20(5):645-674. PMID: 10465518. https://pubmed.ncbi.nlm.nih.gov/10465518/
- Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287(48):40224-40231. PMID: 23038269. PMCID: PMC3504738. https://pubmed.ncbi.nlm.nih.gov/23038269/
- Volkova A, Shadrina M, Kolomin T, Andreeva L, Limborska S, Myasoedov N, Slominsky P. Selank Administration Affects the Expression of Some Genes Involved in GABAergic Neurotransmission. Front Pharmacol. 2016;7:31. PMID: 26924987. PMCID: PMC4757669. https://pmc.ncbi.nlm.nih.gov/articles/PMC4757669/
- Kasian A, Kolomin T, Andreeva L, Bondarenko E, Myasoedov N, Slominsky P, Shadrina M. Peptide Selank Enhances the Effect of Diazepam in Reducing Anxiety in Unpredictable Chronic Mild Stress Conditions in Rats. Behav Neurol. 2017;2017:5091027. PMID: 28280289. PMCID: PMC5322660. https://pmc.ncbi.nlm.nih.gov/articles/PMC5322660/
- Filatova E, Kasian A, Kolomin T, et al. GABA, Selank, and Olanzapine Affect the Expression of Genes Involved in GABAergic Neurotransmission in IMR-32 Cells. Front Pharmacol. 2017;8:89. PMID: 28293190. PMCID: PMC5328971. https://pmc.ncbi.nlm.nih.gov/articles/PMC5328971/
- Zozulya AA, Kost NV, Sokolov OY, et al. The inhibitory effect of Selank on enkephalin-degrading enzymes as a possible mechanism of its anxiolytic activity. Bull Exp Biol Med. 2001;131(4):315-317. PMID: 11550013. https://pubmed.ncbi.nlm.nih.gov/11550013/
- Semenova TP, Kozlovskaia MM, Zuikov AV, et al. Comparison of the effects of selank and tuftsin on the metabolism of serotonin in the brain of rats pretreated with PCPA. Eksp Klin Farmakol. 2009;72(4):6-8. PMID: 19803361. https://pubmed.ncbi.nlm.nih.gov/19803361/
- Inozemtseva LS, Karpenko EA, Dolotov OV, et al. Intranasal administration of the peptide Selank regulates BDNF expression in the rat hippocampus in vivo. Dokl Biol Sci. 2008;421:241-243. PMID: 18841804. doi:10.1134/S0012496608040066. https://pubmed.ncbi.nlm.nih.gov/18841804/
- Zozulia AA, Neznamov GG, Siuniakov TS, et al. Efficacy and possible mechanisms of action of a new peptide anxiolytic selank in the therapy of generalized anxiety disorders and neurasthenia. Zh Nevrol Psikhiatr Im S S Korsakova. 2008;108(4):38-48. PMID: 18454096. https://pubmed.ncbi.nlm.nih.gov/18454096/
- U.S. Food and Drug Administration. Certain Bulk Drug Substances for Use in Compounding That May Present Significant Safety Risks (interim Category 2 / Section 503A bulk drug substances; Selank). https://www.fda.gov/drugs/human-drug-compounding/certain-bulk-drug-substances-use-compounding-may-present-significant-safety-risks
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. Selank is not approved by the U.S. FDA, the European Medicines Agency, or comparable major regulators for the treatment, cure, or prevention of anxiety, generalized anxiety disorder, or any other condition; it is approved and marketed only in Russia, on the basis of a limited evidence base. Its proposed influence on GABA signaling is an open research question, supported by preclinical and small comparator studies and directly contradicted by at least one cell-culture experiment, and is not an established therapeutic mechanism. Nothing here is medical advice or a recommendation for human use. Any legitimate investigation of this compound should occur within properly authorized preclinical or clinical research under appropriate oversight. Readers should consult qualified professionals and applicable regulations before making any decisions.