Few peptides have moved from metabolic research into hepatology as rapidly as semaglutide, a long-acting glucagon-like peptide-1 (GLP-1) receptor agonist whose weight-lowering and insulin-sensitizing effects sit directly upstream of the metabolic drivers of fatty liver disease. The central research question is deceptively simple: if a compound reliably reduces adiposity and improves glycemic control, does it measurably improve the liver histology that defines nonalcoholic steatohepatitis (NASH) — now increasingly termed metabolic dysfunction-associated steatohepatitis (MASH)? Unlike most peptides discussed in a research context, semaglutide has been tested against this question in randomized, biopsy-controlled trials up to phase 3, which lets us move beyond mechanistic speculation into what the histological data actually demonstrate — and, just as importantly, where they stop short.
This article surveys the current evidence with an emphasis on scientific honesty: robust effects on steatosis resolution and inflammation, more modest and later-emerging effects on fibrosis, and a mechanism that is almost entirely indirect. It is written for a scientifically literate audience and is intended as an educational review of the research literature, not as medical guidance.
What Is the Research Question, and Why Has NAFLD Terminology Shifted to MASLD/MASH?
Nonalcoholic fatty liver disease (NAFLD) describes hepatic steatosis — triglyceride accumulation in more than roughly 5% of hepatocytes — in the absence of significant alcohol intake or other secondary causes. Its progressive form, nonalcoholic steatohepatitis (NASH), adds hepatocellular injury (ballooning), lobular inflammation, and, in many cases, progressive fibrosis that can advance to cirrhosis and hepatocellular carcinoma. Because fibrosis stage is the strongest histological predictor of liver-related and all-cause mortality, fibrosis regression — not merely fat clearance — is the endpoint that ultimately matters most in this field.
In 2023, a multi-society Delphi consensus process involving 236 panelists across 56 countries formally revised this nomenclature. NAFLD was replaced by metabolic dysfunction-associated steatotic liver disease (MASLD), and NASH by metabolic dysfunction-associated steatohepatitis (MASH), under an overarching category of steatotic liver disease (SLD).[1] The change was driven by the desire to remove stigmatizing terms (“fatty,” “nonalcoholic”) and to anchor the diagnosis in positive cardiometabolic criteria rather than exclusion. Because much of the semaglutide literature spans this transition, this article uses NAFLD/NASH when citing older trials that used those terms and MASLD/MASH for the more recent phase 3 work, treating the disease constructs as broadly continuous. Readers new to these distinctions may find the peptide and metabolic research glossary a useful reference for the terminology used throughout.
How Might Semaglutide Mechanistically Influence Hepatic Fat Accumulation?
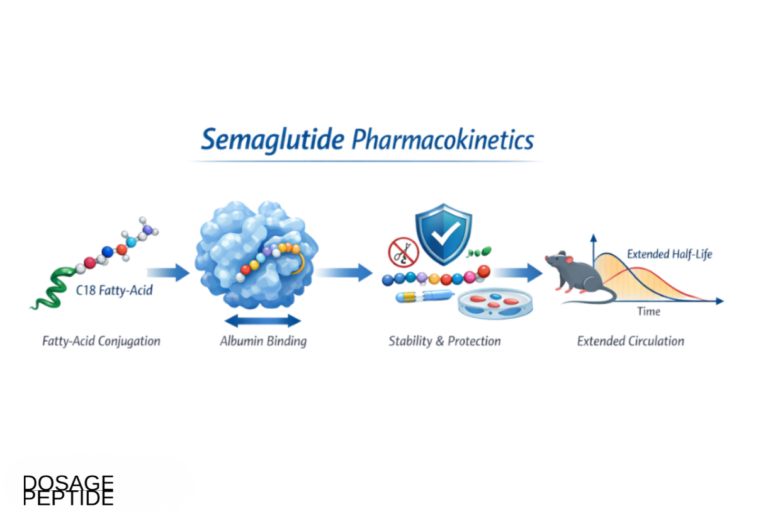
Semaglutide is a GLP-1 receptor agonist engineered for a prolonged half-life (roughly one week) through fatty-acid acylation that promotes albumin binding and resists dipeptidyl peptidase-4 degradation. Its metabolic actions are well characterized: it augments glucose-dependent insulin secretion, suppresses inappropriate glucagon release, slows gastric emptying, and acts centrally to reduce appetite and energy intake. The weight loss and glycemic improvement that follow are substantial — the STEP 1 trial reported a mean body-weight reduction of −14.9% over 68 weeks versus −2.4% with placebo.[2] Since obesity and insulin resistance are the dominant drivers of hepatic steatosis, this magnitude of metabolic change is more than enough, on its own, to plausibly move liver histology.
Is the hepatic effect direct or indirect?
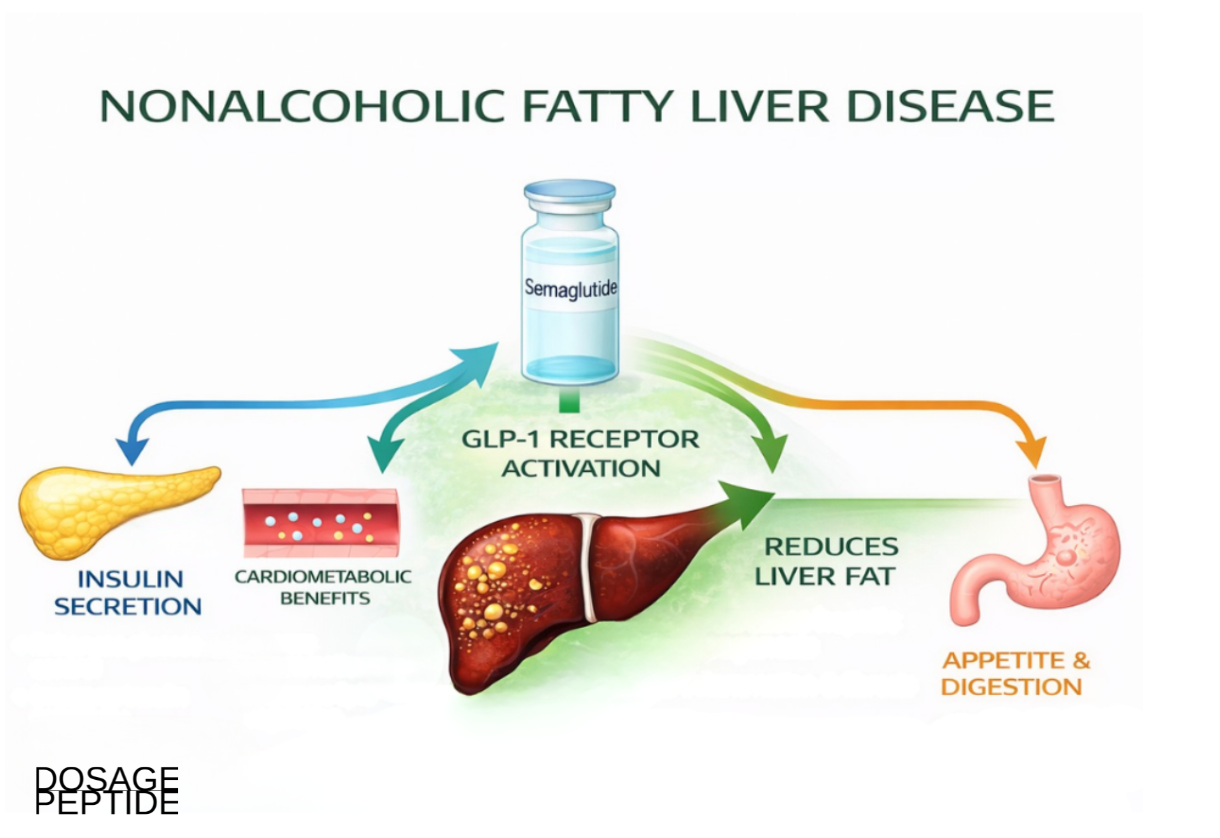
A crucial and frequently misunderstood point: the effect of semaglutide on the liver is almost certainly indirect. Multiple lines of evidence indicate that mature human and rodent hepatocytes do not express functional GLP-1 receptors at meaningful levels, which makes a direct hepatocyte-signaling mechanism unlikely.[3] Instead, the liver appears to benefit through a cascade of upstream systemic changes:
- Weight loss and reduced adipose lipotoxicity. Loss of visceral and ectopic fat lowers the flux of free fatty acids delivered to the liver via the portal circulation, reducing the substrate available for hepatic triglyceride synthesis.
- Improved hepatic and peripheral insulin sensitivity. Better insulin signaling suppresses hepatic gluconeogenesis and blunts de novo lipogenesis, a pathway that is pathologically upregulated in insulin-resistant fatty liver.
- Reduced caloric intake. Central appetite suppression and delayed gastric emptying lower overall energy availability, reinforcing the negative energy balance that clears hepatic fat.
- Secondary anti-inflammatory effects. As metabolic stress and lipotoxicity decline, downstream drivers of hepatocellular injury and lobular inflammation are attenuated — an effect that tracks with, rather than precedes, the metabolic improvement.
Whether any weight-independent or direct component exists remains debated, and some reviews leave the possibility of low-level or context-dependent hepatic signaling open.[4] But the parsimonious and best-supported model is that semaglutide’s hepatic benefit is mediated overwhelmingly through systemic metabolic correction rather than by acting on the hepatocyte itself. This has an important interpretive consequence: hepatic outcomes in trials tend to correlate with the degree of weight loss and glycemic improvement achieved.
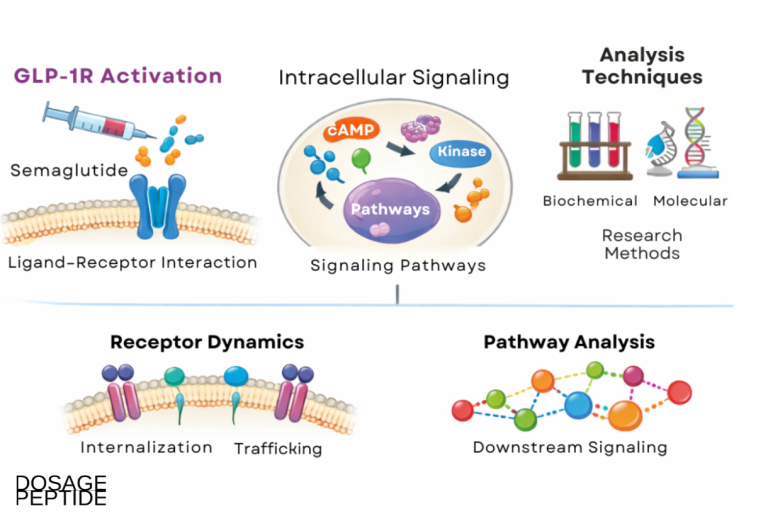
How does GLP-1 receptor signaling actually work?
To understand why semaglutide behaves the way it does — potent metabolically, indirect hepatically — it helps to look at the native incretin biology the drug exploits. Glucagon-like peptide-1 is one of two principal incretin hormones (the other being glucose-dependent insulinotropic polypeptide, GIP). It is secreted by enteroendocrine L cells of the distal small intestine and colon within minutes of nutrient ingestion, and it accounts for a substantial share of the so-called incretin effect: the observation that oral glucose provokes a markedly greater insulin response than an intravenous glucose load producing the same blood-glucose excursion, because the gut-derived hormones amplify beta-cell secretion.[8]
The defining pharmacological feature of this axis is that its insulinotropic action is glucose-dependent. GLP-1 receptor activation on the pancreatic beta cell potentiates glucose-stimulated insulin secretion but has little or no effect when blood glucose is not elevated, which is the mechanistic reason GLP-1 receptor agonists carry a low intrinsic risk of hypoglycemia as monotherapy.[8] The same signaling suppresses inappropriate glucagon release from pancreatic alpha cells and slows gastric emptying, blunting post-prandial glucose spikes.
Receptor distribution explains the breadth of the compound’s effects. The GLP-1 receptor is a class B G-protein-coupled receptor expressed on pancreatic beta cells, but also in the heart (sinoatrial node), vasculature, kidney, gastrointestinal tract, and — critically for appetite — several central nervous system nuclei. Validated receptor sites include the hypothalamic arcuate and paraventricular nuclei and the brainstem’s area postrema and nucleus of the solitary tract, regions that govern satiety, energy balance, and nausea.[8] The area postrema sits at a circumventricular gap in the blood-brain barrier, which helps a large, albumin-bound peptide influence central appetite circuits; it is also the anatomical basis for the class’s characteristic nausea. This central action, reducing food intake, is what couples GLP-1 receptor agonism to meaningful weight loss — and, through weight loss, to the downstream hepatic benefit.
Native GLP-1 is almost useless as a drug because dipeptidyl peptidase-4 (DPP-4) cleaves it within one to two minutes, giving a circulating half-life measured in seconds. Semaglutide’s ~1-week half-life is engineered around this problem through two modifications: substitution at the DPP-4 cleavage site to resist enzymatic degradation, and attachment of a C-18 fatty diacid chain (fatty-acid acylation) via a linker. That lipid tail binds reversibly and with high affinity to circulating albumin, so the vast majority of the drug is sequestered on albumin at any moment, protected from renal filtration and enzymatic attack and released slowly. The result is a pharmacokinetic profile flat enough to support once-weekly dosing — the acylation strategy that separates semaglutide from short-acting predecessors and underlies the sustained metabolic pressure that ultimately moves liver histology.

How Does Semaglutide Affect Inflammation and Fibrotic Pathways in NAFLD/MASH?
Steatohepatitis is distinguished from simple steatosis by active hepatocellular injury and inflammation, which over time recruit hepatic stellate cells and drive the collagen deposition that constitutes fibrosis. In principle, a therapy that reduces steatosis and lipotoxicity should dampen the inflammatory signaling that feeds this cascade — and the clinical data broadly bear this out for the inflammatory component, while telling a more cautious story about fibrosis.
Inflammation and steatohepatitis resolution
Across randomized trials, semaglutide has produced high rates of resolution of steatohepatitis without worsening of fibrosis, a composite histological endpoint that captures the disappearance of ballooning and inflammation. This is where the compound’s signal is strongest and most consistent. Reductions in aminotransferases (ALT, AST) and in imaging-based liver fat (MRI proton density fat fraction) accompany these histological changes, indicating a genuine anti-inflammatory and steatosis-clearing effect rather than a measurement artifact.
Fibrosis: the harder endpoint
Fibrosis regression is biologically slower and mechanistically distinct from resolving inflammation. Established collagen must be remodeled, and this appears to lag behind — and be more difficult to achieve than — the resolution of steatohepatitis. As detailed in the trial-by-trial evidence below, the earliest semaglutide trials failed to demonstrate a statistically significant fibrosis benefit, and it was only in longer, larger phase 3 work that a fibrosis-improvement signal emerged. Even then, the effect is modest in absolute terms. The honest summary is that semaglutide reliably resolves the “hepatitis” of steatohepatitis but affects the “fibrosis” component only partially and later.
What Does the Clinical Evidence Actually Show, by Trial?
Semaglutide’s NAFLD/MASH evidence base is unusually strong for a peptide discussed in research settings, because it rests on randomized, placebo-controlled, biopsy-defined trials rather than surrogate markers alone. The following sections walk through the pivotal studies in order of increasing scale, which also happens to be the order in which the fibrosis picture clarified.
The phase 2 NASH trial (Newsome et al., NEJM 2021)
The foundational study was a 72-week, double-blind, phase 2 randomized trial in patients with biopsy-confirmed NASH and fibrosis stage F1–F3, testing once-daily subcutaneous semaglutide at 0.1, 0.2, or 0.4 mg against placebo.[5] The results defined the field:
| Endpoint | Placebo | Semaglutide 0.1 mg | Semaglutide 0.2 mg | Semaglutide 0.4 mg |
|---|---|---|---|---|
| NASH resolution without worsening fibrosis | 17% | 40% | 36% | 59% (P<0.001 vs placebo) |
| Improvement in fibrosis stage | No statistically significant between-group difference | |||
The 59% NASH-resolution rate at the highest dose was, at the time, the highest response an active agent had achieved in a NASH trial — a landmark finding. Yet the trial explicitly did not show a significant improvement in fibrosis stage. This dissociation, high resolution of steatohepatitis alongside a null fibrosis result, established the recurring theme of the semaglutide hepatology literature and set expectations that fibrosis benefit, if achievable, would require longer exposure or larger samples.
The NASH-related cirrhosis trial (Loomba et al., Lancet Gastroenterology & Hepatology 2023)
A subsequent phase 2 trial extended the question to a more advanced population: 71 patients with biopsy-confirmed NASH-related (compensated) cirrhosis received once-weekly semaglutide 2.4 mg or placebo for 48 weeks.[6] The primary endpoint — improvement in fibrosis without worsening of NASH — was not met: fibrosis improved in 11% of the semaglutide group versus 29% of the placebo group, a non-significant difference. Nor did the trial show a significant benefit for NASH resolution in this cirrhotic cohort. What semaglutide did deliver were the expected cardiometabolic improvements — weight loss, better glycemic control, improved lipids, and reduced liver fat on imaging.
The interpretation is important and sobering: once fibrosis has advanced to cirrhosis, the metabolic correction semaglutide provides may be insufficient to reverse the established structural disease within a one-year window. This reinforced the view that semaglutide’s hepatic value is greatest earlier in the disease course, before fibrosis becomes fixed.
The phase 3 ESSENCE trial (NEJM 2025)
The most consequential evidence to date comes from the ESSENCE trial, an ongoing phase 3, multicenter, randomized, double-blind, placebo-controlled study that assigned 1,197 patients with biopsy-defined MASH and fibrosis stage F2 or F3 in a 2:1 ratio to once-weekly subcutaneous semaglutide 2.4 mg or placebo, with a planned 240-week duration. The pre-specified 72-week interim histology analysis (part 1) was published in the New England Journal of Medicine in 2025.[7] For the first time in a phase 3 setting, both key endpoints were met:
| Histological endpoint at 72 weeks | Semaglutide 2.4 mg (n=534) | Placebo (n=266) |
|---|---|---|
| Resolution of steatohepatitis without worsening of fibrosis | 62.9% | 34.3% |
| Improvement in liver fibrosis without worsening of steatohepatitis | 36.8% | 22.4% |
| Both resolution of steatohepatitis and fibrosis improvement | 32.7% | 16.1% |
Two things stand out. First, the steatohepatitis-resolution result (62.9% vs 34.3%) confirms and extends the strong anti-inflammatory signal seen since the phase 2 work. Second — and this is the genuinely new finding — the fibrosis-improvement endpoint was met: 36.8% versus 22.4%. This is the first time semaglutide has demonstrated a statistically significant fibrosis benefit in a biopsy-controlled trial, and it appeared only in a large phase 3 sample with F2–F3 (pre-cirrhotic) fibrosis over a longer interval.
The appropriate reading is measured optimism rather than triumphalism. The fibrosis effect, while real, is modest: roughly a 14-percentage-point absolute advantage over placebo, with the majority of treated patients not achieving fibrosis regression at 72 weeks. The trial enrolled specifically F2–F3 patients — the population most likely to respond — and the results do not extend to cirrhosis, where the Loomba trial found no benefit. The safety profile in ESSENCE was consistent with prior semaglutide experience, dominated by the expected gastrointestinal effects. Longer-term outcomes from the full 240-week study, including whether histological improvement translates into reduced progression to cirrhosis and clinical events, remain to be reported.
Putting the trials together
Read as a sequence, the trials tell a coherent story. Semaglutide’s effect on steatosis and steatohepatitis inflammation is robust and reproducible across doses and populations. Its effect on fibrosis is stage-dependent and time-dependent: absent in the shorter phase 2 studies, absent in established cirrhosis, but present and modest in a large F2–F3 phase 3 cohort followed for 72 weeks. The indirect, metabolically driven mechanism outlined earlier fits this stage- and time-dependent pattern well.
How Does Semaglutide Compare With Other Agents in the MASH Landscape?
Semaglutide does not exist in a vacuum. MASH pharmacotherapy went from having zero approved drugs to a rapidly maturing pipeline in the span of a few years, and interpreting semaglutide’s data honestly requires situating it against the agents it will be measured against. These fall into several mechanistic classes, and — importantly — the trials differ in length, fibrosis stage enrolled, and endpoint definitions, so cross-trial numbers should be read as orientation rather than head-to-head comparison. No large randomized trial has directly pitted these agents against one another.
Resmetirom: the first approved MASH drug
Resmetirom (Rezdiffra) is an oral, liver-directed thyroid hormone receptor-β (THR-β) selective agonist. In hepatocytes, THR-β activation upregulates mitochondrial fatty-acid oxidation and lowers intrahepatic lipid — a mechanism that, unlike semaglutide’s, is directly hepatic. The phase 3 MAESTRO-NASH trial randomized 966 patients with F1B–F3 fibrosis to resmetirom 80 mg, 100 mg, or placebo, with a 52-week biopsy.[9] NASH resolution without worsening of fibrosis occurred in 25.9% (80 mg) and 29.9% (100 mg) versus 9.7% with placebo, and fibrosis improvement of at least one stage in 24.2% and 25.9% versus 14.2%. In March 2024 resmetirom became the first FDA-approved therapy for MASH with fibrosis, a milestone that reframed the field: any new agent, semaglutide included, is now evaluated against an existing standard rather than against placebo alone.
Tirzepatide: dual GIP/GLP-1 agonism
Tirzepatide is a once-weekly dual agonist of the GIP and GLP-1 receptors — engaging both incretin axes described earlier — and produces even greater weight loss than semaglutide in obesity trials. The phase 2 SYNERGY-NASH trial randomized 190 patients with F2–F3 MASH to tirzepatide 5, 10, or 15 mg or placebo for 52 weeks.[10] Absence of MASH without worsening of fibrosis was achieved in 51.8%, 62.8%, and 73.3% across ascending doses versus 13.2% with placebo. The secondary fibrosis endpoint was also met: at least one-stage fibrosis improvement without worsening of MASH occurred in 59.1%, 53.3%, and 54.2% across doses versus 32.8% with placebo — every dose roughly doubling the placebo rate, a genuine fibrosis signal comparable in absolute terms to semaglutide’s ESSENCE result. The salient distinction is therefore one of evidence maturity, not effect: tirzepatide’s histology remains a smaller phase 2 secondary read-out versus semaglutide’s completed phase 3.
Survodutide: glucagon/GLP-1 co-agonism
Survodutide is a dual glucagon-receptor/GLP-1-receptor agonist; the added glucagon-receptor arm is hypothesized to increase hepatic lipid oxidation and energy expenditure directly, complementing GLP-1-driven weight loss. Its 48-week phase 2 trial enrolled F1–F3 MASH patients on 2.4, 4.8, or 6.0 mg or placebo.[11] Improvement in MASH without worsening of fibrosis occurred in 47%, 62%, and 43% across doses versus 14% with placebo, and roughly 34% achieved at least one-stage fibrosis improvement (6.0 mg) versus 22% with placebo — a suggestive rather than definitive fibrosis result that the authors framed as supporting further phase 3 investigation. Gastrointestinal adverse effects were prominent (nausea up to 66%).
The older and adjacent classes
Two agents predate the incretin era and remain historically important. The PIVENS trial (2010) randomized 247 non-diabetic NASH patients to pioglitazone 30 mg, vitamin E 800 IU, or placebo for 96 weeks.[12] Vitamin E significantly improved steatosis, inflammation, and ballooning versus placebo; pioglitazone improved many features but caused weight gain. Crucially, neither improved fibrosis — an early and enduring lesson that metabolic and anti-inflammatory improvement does not automatically translate into fibrosis regression, exactly the pattern semaglutide later reproduced.
Several agents work through nuclear-receptor or growth-factor pathways rather than incretin biology. Lanifibranor, a pan-PPAR (α/δ/γ) agonist, in the phase 2b NATIVE trial produced NASH resolution without worsening fibrosis in 49% (1200 mg) versus 22% placebo and fibrosis improvement in 48% versus 29%.[13] FGF21 analogs — efruxifermin and pegozafermin — mimic the metabolic hormone FGF21 to improve insulin sensitivity and lipid handling; efruxifermin’s phase 2b HARMONY trial reported significant fibrosis improvement over placebo in F2–F3 patients.[14] These classes are attractive precisely because several act on the hepatocyte and stellate cell more directly than GLP-1 agonism does, and combination strategies (e.g., an incretin agent plus an FGF21 analog) are an active research direction.
| Agent | Class / mechanism | Highest trial stage (MASH) | Steatohepatitis resolution | Fibrosis improvement | Hepatic action |
|---|---|---|---|---|---|
| Semaglutide | GLP-1 receptor agonist | Phase 3 (ESSENCE, 72-wk interim) | 62.9% vs 34.3% | 36.8% vs 22.4% (F2–F3) | Indirect (metabolic) |
| Resmetirom | THR-β agonist (oral) | Phase 3 (MAESTRO-NASH); FDA-approved | 25.9–29.9% vs 9.7% | 24.2–25.9% vs 14.2% | Direct (hepatocyte) |
| Tirzepatide | Dual GIP/GLP-1 agonist | Phase 2 (SYNERGY-NASH) | 51.8–73.3% vs 13.2% | 53.3–59.1% vs 32.8% | Indirect (metabolic) |
| Survodutide | Glucagon/GLP-1 co-agonist | Phase 2 | 47–62% vs 14% | ~34% vs 22% | Mixed (metabolic + hepatic) |
| Lanifibranor | Pan-PPAR agonist | Phase 2b (NATIVE) | 49% vs 22% | 48% vs 29% | Direct (multi-tissue) |
| Efruxifermin | FGF21 analog | Phase 2b (HARMONY) | Significant vs placebo | Significant vs placebo | Direct (metabolic/hepatic) |
| Pioglitazone / Vitamin E | PPAR-γ / antioxidant | Phase 3 (PIVENS) | Improved (vit E) | No improvement | Direct (hepatocyte) |
The pattern across the table is instructive. Agents that act directly on the liver (resmetirom, FGF21 analogs, lanifibranor) tend to show cleaner fibrosis signals earlier, whereas the incretin-based agents (semaglutide, tirzepatide) generate their most dramatic numbers on resolution and win the fibrosis endpoint more gradually, consistent with the indirect mechanism. Semaglutide’s distinctive advantage is not a uniquely large hepatic effect but the maturity of its evidence — a completed, positive phase 3 histology read-out — combined with a systemic metabolic and cardiovascular benefit the liver-directed agents do not provide.
What about cardiovascular and broader metabolic outcomes?
The MASH population is, by definition, a high-cardiometabolic-risk population: these patients carry the obesity, insulin resistance, dyslipidemia, and hypertension that also drive atherosclerotic cardiovascular disease, and cardiovascular events — not liver failure — are the leading cause of death in most people with MASLD. An agent’s value in this setting therefore depends partly on effects reaching beyond the liver. This is where semaglutide’s broader profile becomes relevant.
The SELECT trial (2023) was a large, multicenter, randomized, placebo-controlled cardiovascular-outcomes study of once-weekly semaglutide 2.4 mg in adults with overweight or obesity and established cardiovascular disease but without diabetes.[15] Over a mean follow-up of about 40 months, semaglutide reduced the primary composite of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke (MACE) by roughly 20% versus placebo. Prespecified analyses indicated the cardiovascular benefit was only partly explained by weight loss, hinting at additional mechanisms.
SELECT did not enroll or measure MASH, so it is not liver-outcome evidence. Its relevance is contextual but substantial: the same weekly 2.4 mg regimen studied in ESSENCE independently lowers hard cardiovascular events in precisely the comorbid, high-risk phenotype that populates MASH clinics. For a disease whose patients are more likely to die of a heart attack than of cirrhosis, a therapy that simultaneously improves liver histology and reduces cardiovascular risk occupies a different position than a purely liver-directed drug — a point in semaglutide’s favor that the histology tables alone do not capture. It remains, however, a separate line of evidence: cardiovascular benefit in one trial population does not establish hepatic benefit, and vice versa.
Why the mechanistic differences point toward combinations
The comparison above exposes a complementarity that is shaping the next wave of MASH research. Semaglutide and other incretin agents drive the upstream metabolic correction — weight, glycemia, lipotoxicity — that clears steatosis and resolves inflammation, but they reach the fibrotic compartment only indirectly and slowly. Liver-directed agents such as resmetirom and the FGF21 analogs act closer to the hepatocyte and stellate cell, producing cleaner early fibrosis signals but without the systemic weight and cardiovascular effects. In principle, pairing an incretin agent that removes the metabolic driver with a liver-targeted agent that accelerates fibrosis remodeling could address both compartments at once, and early combination studies — for example, an FGF21 analog combined with a GLP-1 receptor agonist in MASH with type 2 diabetes — have begun testing exactly this logic.[16] Whether combinations deliver additive histological benefit without additive toxicity is unresolved, but the mechanistic rationale is coherent and is a reason the field is unlikely to settle on any single agent as a universal answer.
The practical takeaway from the landscape is that semaglutide is best understood not as the “best” MASH drug in isolation but as the incretin backbone with the most mature evidence and a uniquely broad metabolic-cardiovascular footprint. Its position will keep shifting as tirzepatide and survodutide report phase 3 histology, as combination data mature, and as the first real-world experience with resmetirom accumulates.
Which Research Models and Methods Are Used to Study Semaglutide in Fatty Liver Disease?
Understanding how these findings are generated helps calibrate their weight. NAFLD/MASH research spans several model systems, each with characteristic strengths and limitations.
Preclinical and in vitro models
Rodent models of diet-induced steatohepatitis (high-fat, high-fructose, or methionine-choline-deficient diets) and genetic obesity models (ob/ob, db/db) are used to probe mechanism and dose-response before clinical testing. These systems allow controlled interrogation of hepatic lipid handling, inflammatory signaling, and stellate-cell activation, and they informed early hypotheses about GLP-1 receptor agonists in the liver. Their principal limitation is imperfect fidelity to human MASH histology and to the human fibrosis timeline, which is why favorable rodent fibrosis data must always be validated in biopsy-controlled human trials. The absence of hepatocyte GLP-1 receptors also means cell-culture systems cannot straightforwardly model the indirect, systemically mediated mechanism at play in vivo.
Clinical trial methodology and endpoints
Human MASH trials are methodologically demanding. The reference-standard endpoints — resolution of steatohepatitis and change in fibrosis stage — require paired liver biopsies read by blinded pathologists, an invasive and variable procedure. Biopsy sampling error and inter-reader variability inject noise that can obscure modest effects, which is one reason large sample sizes (as in ESSENCE) are needed to detect a fibrosis signal. Non-invasive markers help supplement biopsy: MRI-PDFF quantifies liver fat with high sensitivity, transient elastography and MR elastography estimate stiffness as a fibrosis surrogate, and serum panels (e.g., ELF, FIB-4) track fibrogenesis. In research contexts these tools are increasingly used to enrich cohorts and monitor response, though regulatory histological endpoints still anchor pivotal trials.
Several structural features of MASH trials help explain why the semaglutide fibrosis story took so long to resolve and why cross-trial comparison is treacherous. First, the placebo response in these studies is unusually high — often 20–35% for steatohepatitis resolution and 14–25% for fibrosis improvement, as the tables above show — because trial enrollment itself prompts lifestyle change and because sampling variability on a second biopsy can register apparent “improvement” that is partly regression to the mean. A high placebo rate raises the bar an active drug must clear and demands larger samples. Second, the histological endpoints are semi-quantitative categorical scores (for example, NAS components and fibrosis stage), so a one-stage change can mean very different biological things in different patients. Third, the fibrosis endpoint is intrinsically slow: because collagen turnover takes time, a 24- or 48-week study may simply be too short to reveal a remodeling effect that a 72-week study captures — one likely reason the phase 2 semaglutide trials were null on fibrosis while phase 3 ESSENCE was positive. Enrichment for F2–F3 disease, where fibrosis is active but not yet fixed, further concentrates the population most able to respond. Recognizing these design forces is essential to reading any single trial’s numbers without over- or under-interpreting them.
What Is the Research Handling, Reconstitution, and Dosing Context?
Important framing: the material in this section is provided for laboratory and educational reference only. Semaglutide sold as a research compound is not approved for human use in that context, and nothing here constitutes medical advice or a recommendation for administration. The therapeutic dosing figures cited above (for example, once-weekly 2.4 mg in ESSENCE and STEP 1, or once-daily 0.4 mg in the phase 2 NASH trial) are reported strictly to describe what the published trials tested, not to guide any use.
In research settings, semaglutide is typically supplied as a lyophilized powder that must be reconstituted with bacteriostatic water before handling in experimental workflows. Accurate reconstitution is a common source of methodological error: the final concentration depends on both the peptide mass in the vial and the diluent volume added, and small volumetric mistakes translate directly into large errors in the intended per-unit quantity. Investigators working through these calculations may find the peptide reconstitution guide and the reconstitution and dosage calculator useful for verifying concentration math and syringe-unit conversions before any experimental procedure.
For structured, vial-size-specific handling references, the semaglutide 5 mg vial dosage protocol documents reconstitution parameters and escalation frameworks used in research contexts, with a parallel reference for the semaglutide 10 mg vial. Because dual-incretin and amylin-based strategies are an active area of comparative research, a combined cagrilintide-semaglutide blend protocol reference is also available for investigators studying co-agonist approaches. In all cases, cold-chain storage, protection from light, and avoidance of freeze-thaw cycling and vigorous agitation are standard practices for preserving peptide integrity, since degradation or aggregation can confound experimental readouts.
What Are the Key Limitations and Open Questions?
Even with phase 3 data in hand, several important caveats bound what can honestly be claimed.
Fibrosis regression is modest and stage-limited
The ESSENCE fibrosis benefit, though statistically significant, left most treated patients without fibrosis improvement at 72 weeks, and it applied to F2–F3 disease only. In established cirrhosis, the Loomba trial found no fibrosis benefit. Whether semaglutide can meaningfully alter the long-term trajectory toward cirrhosis and hard clinical outcomes (decompensation, hepatocellular carcinoma, liver-related death) is not yet established and awaits the full ESSENCE follow-up.
Benefit appears tied to sustained metabolic control
Because the hepatic effect is indirect and proportional to weight and glycemic improvement, it is mechanistically expected that discontinuation and subsequent weight regain would erode liver benefits. Weight regain after cessation is well documented in the obesity literature for this class, raising the durability question of whether histological gains persist off-treatment — a question the current biopsy trials, which measure on-treatment histology, do not directly answer.
Gastrointestinal tolerability and adherence
The dominant adverse-effect signal across the semaglutide program is gastrointestinal — nausea, vomiting, diarrhea, and constipation, concentrated during dose escalation. In trials these effects are common but usually mild-to-moderate and transient; nonetheless, they influence adherence and dropout, which can complicate the interpretation of long studies. In the ESSENCE interim analysis the safety profile was consistent with prior semaglutide experience, with no unexpected signals.
Methodological and translational limits
Biopsy-based endpoints carry sampling and reader variability; trial populations are enriched and may not represent the full spectrum of real-world MASLD; and the shift in nomenclature from NAFLD/NASH to MASLD/MASH, while conceptually continuous, means older and newer trials used slightly different diagnostic framings. Finally, semaglutide is one agent within a rapidly evolving landscape that now includes dual GIP/GLP-1 and triple agonists and resmetirom (a thyroid-hormone-receptor-β agonist approved for MASH), so its relative position will continue to shift as comparative data mature.
What Safety and Handling Considerations Apply in Research Use?
In a research context, safety and handling considerations center on material integrity and laboratory practice rather than clinical administration. Peptide purity and batch-to-batch consistency directly affect the reproducibility of any experimental readout: impurities, incomplete synthesis products, or aggregation can shift apparent activity and confound comparisons across experiments. Analytical characterization (for example, HPLC purity assessment and mass-spectrometric identity confirmation) is therefore foundational to reliable work.
Standard handling practice includes reconstitution with appropriate diluent under aseptic technique, storage of lyophilized material and reconstituted solution at recommended temperatures, protection from light, and avoidance of repeated freeze-thaw cycles that promote degradation. Because semaglutide research compounds are not intended or approved for human use, they should be handled strictly within institutional and jurisdictional guidelines for laboratory materials.
Frequently Asked Questions
Does semaglutide reduce liver fat in NAFLD/MASH research?
Yes. Across randomized, biopsy-controlled trials, semaglutide consistently reduces hepatic steatosis and produces high rates of steatohepatitis resolution. In the phase 2 NASH trial, up to 59% of patients achieved NASH resolution without worsening fibrosis, and the phase 3 ESSENCE trial reported 62.9% resolution versus 34.3% with placebo. Imaging measures such as MRI-PDFF and liver enzymes (ALT, AST) improve in parallel, reflecting genuine fat clearance and reduced inflammation.
Does semaglutide reverse liver fibrosis?
Only partially, and only in earlier-stage disease. The phase 2 NASH trial and the NASH-cirrhosis trial did not show a significant fibrosis benefit. The larger phase 3 ESSENCE trial was the first to demonstrate a significant fibrosis improvement (36.8% versus 22.4% with placebo) in F2–F3 patients at 72 weeks, but the effect is modest and most treated patients did not achieve fibrosis regression. In established cirrhosis, no fibrosis benefit has been shown.
How does semaglutide affect the liver if hepatocytes lack GLP-1 receptors?
The hepatic effect is indirect. Mature hepatocytes do not express functional GLP-1 receptors at meaningful levels, so semaglutide is thought to improve the liver through systemic changes: weight loss that reduces adipose lipotoxicity and portal free-fatty-acid flux, improved hepatic and peripheral insulin sensitivity that lowers de novo lipogenesis, and reduced caloric intake. Hepatic outcomes therefore tend to track the degree of metabolic and weight improvement achieved.
What is the difference between NAFLD, NASH, MASLD, and MASH?
NAFLD (now MASLD) refers to fat accumulation in the liver without significant alcohol intake; NASH (now MASH) is its inflammatory, injurious form that can progress to fibrosis and cirrhosis. In 2023 a multi-society Delphi consensus replaced the older terms to remove stigma and anchor diagnosis in cardiometabolic criteria. The disease constructs are broadly continuous, so older semaglutide trials (NASH) and newer ones (MASH) address the same underlying condition.
Is semaglutide approved for treating fatty liver disease?
Semaglutide has robust clinical trial evidence in MASH, culminating in the positive phase 3 ESSENCE interim results, but readers should note that regulatory status varies by product, indication, and jurisdiction and changes over time. Research-grade semaglutide sold for laboratory use is not approved for human use in that context. This article is educational and does not provide medical advice; questions about approved therapies should be directed to qualified clinicians.
Why do the effects on inflammation appear stronger than on fibrosis?
Resolving steatohepatitis means clearing fat, hepatocellular ballooning, and lobular inflammation — processes that respond relatively quickly to metabolic correction. Fibrosis reflects accumulated collagen that must be actively remodeled, which is biologically slower and harder to reverse. This is why a strong steatohepatitis-resolution signal appeared early (phase 2), while a significant fibrosis signal emerged only in the larger, longer phase 3 trial, and only in pre-cirrhotic disease.
How is semaglutide studied in research models?
Preclinical work uses diet-induced and genetic rodent models of steatohepatitis to probe mechanism, while pivotal human evidence relies on randomized, placebo-controlled trials with paired liver biopsies read by blinded pathologists. Non-invasive tools — MRI-PDFF for fat, elastography for stiffness, and serum panels such as FIB-4 and ELF for fibrogenesis — supplement biopsy for cohort enrichment and monitoring. Large samples are needed because biopsy sampling and reader variability can obscure modest effects.
How does semaglutide compare with resmetirom for MASH?
They differ mechanistically and in evidence type. Resmetirom (Rezdiffra) is an oral, liver-directed THR-β agonist that acts directly on hepatocytes and, in the phase 3 MAESTRO-NASH trial, achieved NASH resolution in about 26–30% and fibrosis improvement in about 24–26% versus placebo — making it the first FDA-approved MASH therapy. Semaglutide acts indirectly through systemic metabolic correction and posted higher resolution numbers in ESSENCE (62.9%) with a modest fibrosis benefit (36.8% vs 22.4%). Because the trials used different populations, endpoints, and durations, these figures orient rather than establish superiority; no head-to-head trial exists. Semaglutide additionally carries cardiovascular-outcome data that resmetirom does not.
How does semaglutide compare with tirzepatide for MASH?
Tirzepatide is a dual GIP/GLP-1 receptor agonist that produces greater weight loss than semaglutide and, in the phase 2 SYNERGY-NASH trial, achieved absence of MASH without worsening fibrosis in up to 73.3% versus 13.2% with placebo at the highest dose. Those resolution numbers are striking, but tirzepatide’s MASH evidence remains phase 2, whereas semaglutide has completed positive phase 3 histology (ESSENCE). SYNERGY-NASH also met its secondary fibrosis endpoint (about 53–59% one-stage improvement versus 33% with placebo), so the difference is one of evidence maturity — phase 2 versus phase 3 — rather than a weaker fibrosis effect. The two are best viewed as leading incretin-based candidates at different stages of validation rather than as established superior/inferior.
Do liver-histology improvements last after stopping semaglutide?
This is not directly answered by the current biopsy trials, which measure on-treatment histology. Because the hepatic benefit is indirect and proportional to weight and glycemic control, and because weight regain after discontinuation is well documented for this drug class, it is mechanistically expected that stopping treatment could erode histological gains as metabolic parameters revert. Whether any improvement persists off-treatment — and for how long — remains an open research question. This durability uncertainty is one reason MASH is generally framed as a chronic condition requiring sustained rather than time-limited intervention.
What role do non-invasive biomarkers play versus biopsy?
Liver biopsy remains the regulatory reference standard for MASH trial endpoints, but it is invasive and subject to sampling and reader variability. Non-invasive biomarkers increasingly supplement it: MRI-PDFF quantifies liver fat, vibration-controlled transient elastography and MR elastography estimate stiffness as a fibrosis surrogate, and serum panels such as FIB-4 and the Enhanced Liver Fibrosis (ELF) score track fibrogenesis. In research these tools enrich cohorts, monitor response, and reduce the number of biopsies needed, though pivotal efficacy claims still rest on paired histology. Their growing validation is gradually shifting how MASH trials are designed.
References
- Rinella ME, Lazarus JV, Ratziu V, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Journal of Hepatology. 2023.
- Wilding JPH, Batterham RL, Calanna S, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity (STEP 1). New England Journal of Medicine. 2021;384(11):989–1002.
- Patel Chavez C, Cusi K, Kadiyala S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. The Journal of Clinical Endocrinology & Metabolism. 2022;107(1):29–38.
- Abdelmalek MF, Harrison SA, Sanyal AJ. The Emerging Role of Glucagon-Like Peptide-1 Receptor Agonists for the Treatment of Metabolic Dysfunction-Associated Steatohepatitis. Clinical Gastroenterology and Hepatology. 2024.
- Newsome PN, Buchholtz K, Cusi K, et al. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. New England Journal of Medicine. 2021;384(12):1113–1124.
- Loomba R, Abdelmalek MF, Armstrong MJ, et al. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: a randomised, placebo-controlled phase 2 trial. The Lancet Gastroenterology & Hepatology. 2023;8(6):511–522.
- Sanyal AJ, Newsome PN, Kliers I, et al. Phase 3 Trial of Semaglutide in Metabolic Dysfunction–Associated Steatohepatitis (ESSENCE). New England Journal of Medicine. 2025;392(21):2089–2099.
- Baggio LL, Drucker DJ. Biology of Incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157.
- Harrison SA, Bedossa P, Guy CD, et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis (MAESTRO-NASH). New England Journal of Medicine. 2024;390(6):497–509.
- Loomba R, Hartman ML, Lawitz EJ, et al. Tirzepatide for Metabolic Dysfunction–Associated Steatohepatitis with Liver Fibrosis (SYNERGY-NASH). New England Journal of Medicine. 2024;391(4):299–310.
- Sanyal AJ, Bedossa P, Fraessdorf M, et al. A Phase 2 Randomized Trial of Survodutide in MASH and Fibrosis. New England Journal of Medicine. 2024;391(4):311–319.
- Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis (PIVENS). New England Journal of Medicine. 2010;362(18):1675–1685.
- Francque SM, Bedossa P, Ratziu V, et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH (NATIVE). New England Journal of Medicine. 2021;385(17):1547–1558.
- Harrison SA, Frias JP, Neff G, et al. Safety and efficacy of once-weekly efruxifermin versus placebo in non-alcoholic steatohepatitis (HARMONY): a phase 2b trial. The Lancet Gastroenterology & Hepatology. 2023;8(12):1080–1093.
- Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes (SELECT). New England Journal of Medicine. 2023;389(24):2221–2232.
- Harrison SA, Frias JP, Lucas KJ, et al. Safety and Efficacy of Efruxifermin in Combination With a GLP-1 Receptor Agonist in Patients With NASH/MASH and Type 2 Diabetes in a Randomized Phase 2 Study. Clinical Gastroenterology and Hepatology. 2024.