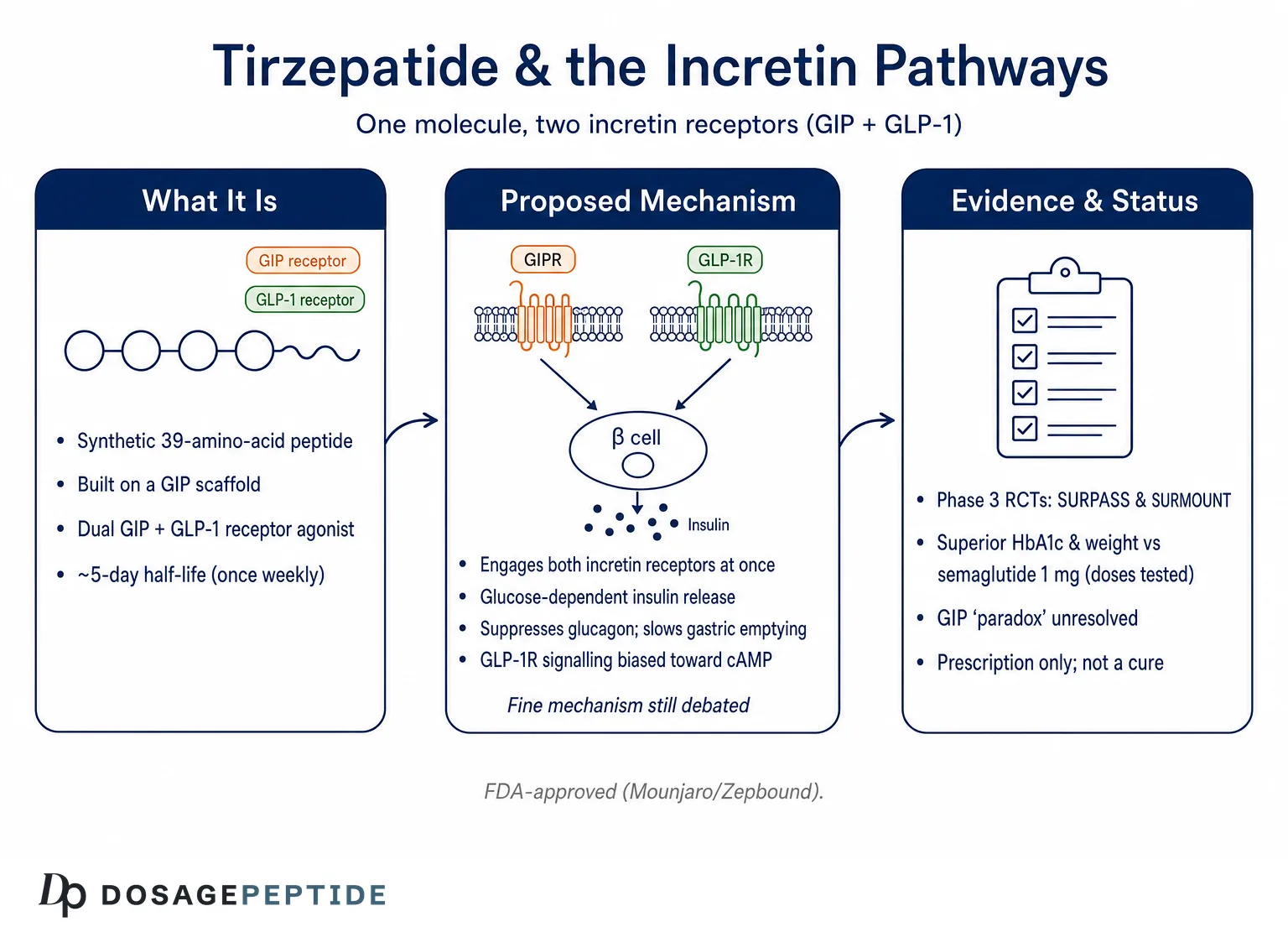
Few molecules have reshaped the conversation around metabolic pharmacology as quickly as tirzepatide. Marketed as Mounjaro for type 2 diabetes and Zepbound for chronic weight management, it is the first medicine designed from the ground up to engage two incretin receptors at once: the receptor for glucose-dependent insulinotropic polypeptide (GIP) and the receptor for glucagon-like peptide-1 (GLP-1).1 The question posed in this article—how does tirzepatide influence incretin pathways?—is not rhetorical. It sits at the center of an active scientific debate about why activating two related but distinct hormone systems produces metabolic effects that appear to exceed what either pathway delivers alone.
This page is an educational overview written for readers who want to understand the biology rather than a prescribing guide. Tirzepatide is an approved therapeutic, and that fact matters: unlike many compounds discussed in the research-peptide community, its mechanism and clinical profile rest on a large, peer-reviewed evidence base including multiple phase 3 randomized controlled trials.4,5 At the same time, the “how” of tirzepatide—the molecular choreography by which it binds, signals, and biases each receptor—remains an area where honest scientists still disagree. We will separate what is well established from what is genuinely unsettled.
Throughout, the goal is mechanistic precision without hype. Tirzepatide does not “cure” diabetes or obesity; it modifies physiology in ways that, in trials, improved glycemic and weight-related outcomes for many participants while carrying a real side-effect burden. Understanding the incretin pathways it recruits is the best foundation for interpreting those results critically, and for appreciating why the next generation of dual and triple agonists is being built the way it is.
What Tirzepatide Is and Where It Came From
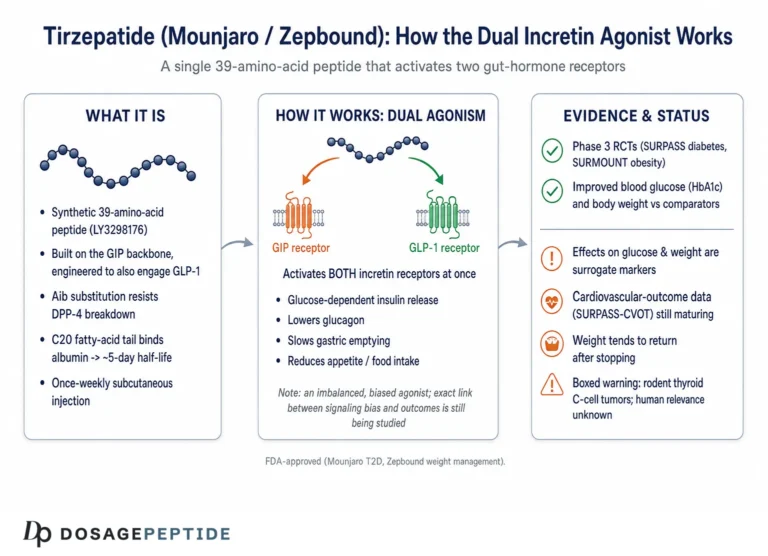
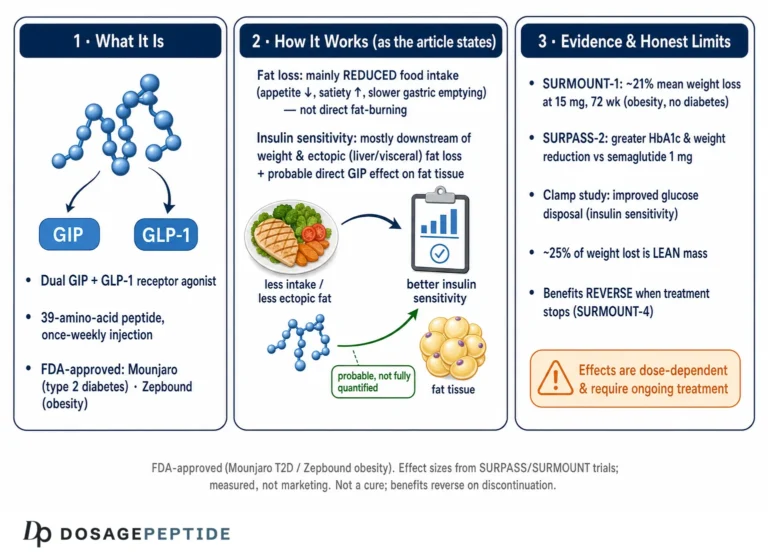
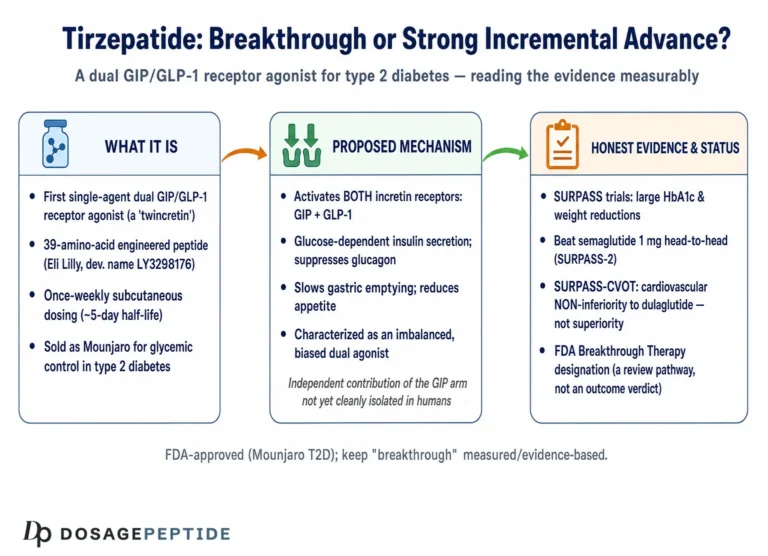
Tirzepatide (development code LY3298176) is a synthetic 39-amino-acid peptide engineered by Eli Lilly. Its backbone is based on the sequence of native GIP rather than GLP-1, which is a design choice with mechanistic consequences we will return to.1,2 Onto that GIP-derived scaffold, the developers grafted structural elements that confer meaningful GLP-1 receptor activity, producing a single molecule that behaves as a dual, or “twincretin,” agonist. A C20 fatty diacid moiety is attached through a linker at a specific lysine residue; this lipidation binds albumin in the circulation, slowing clearance and extending the half-life to roughly 5 days so that once-weekly subcutaneous dosing maintains steady exposure.7,9
The rationale for combining the two incretins grew out of decades of work on the incretin effect. Clinicians observed that oral glucose triggers a far larger insulin response than an intravenous glucose load producing the same blood-sugar level—evidence that the gut releases insulinotropic signals during digestion.6 Two hormones account for most of that effect: GIP, whose sequence was characterized in 1971 and which is secreted by intestinal K-cells, and GLP-1, identified in the 1980s and secreted by L-cells.6 GLP-1 receptor agonists such as exenatide, liraglutide, dulaglutide, and semaglutide became successful diabetes and obesity drugs. GIP, by contrast, had a more ambiguous reputation, and its therapeutic value was for years considered doubtful in type 2 diabetes because its insulinotropic action appears blunted in that condition.6
Tirzepatide was the bet that adding GIP-receptor engagement to GLP-1 agonism—rather than abandoning GIP—could unlock additive or synergistic benefit. Preclinical and early-phase work suggested that it did, with effects on glucose and body weight that exceeded selective GLP-1 receptor agonists in head-to-head diabetes trials.4 The compound advanced through the SURPASS program in type 2 diabetes and the SURMOUNT program in obesity, and the U.S. Food and Drug Administration approved it as Mounjaro in May 2022 and as Zepbound in November 2023.8,13 For readers cataloging the compound alongside its relatives, our reference pages on the tirzepatide protocol and the broader peptide glossary place it in context with other incretin agents.
The naming convention around tirzepatide is worth clarifying because it is a frequent source of confusion. “Tirzepatide” is the international nonproprietary name for the molecule itself—a single active pharmaceutical ingredient. “Mounjaro” and “Zepbound” are brand names for products containing that same molecule, differing chiefly in their approved indications and, to a degree, in dosing presentation rather than in the peptide they deliver. This mirrors the pattern seen with semaglutide, which is sold as Ozempic and Rybelsus for diabetes and as Wegovy for weight management. Recognizing that one molecule can wear several commercial names helps a reader avoid the mistake of treating the brands as pharmacologically distinct agents when they are, at the level of incretin pharmacology, the same compound acting through the same two receptors.
It is also historically instructive that tirzepatide emerged from a specific lineage of medicinal chemistry rather than appearing in isolation. The concept of unimolecular multi-agonists—single peptides engineered to hit two or more metabolically relevant receptors—had been explored in academic laboratories for more than a decade before tirzepatide reached patients, including early GIP/GLP-1 co-agonists and GLP-1/glucagon co-agonists tested in animal models. Tirzepatide was the first such molecule to clear the full gauntlet of phase 3 trials and regulatory review, but it is best understood as the leading edge of a broader research program that continues today with triple agonists and other combinations. That lineage is why the mechanistic questions raised by tirzepatide—especially about the role of GIP—carry weight far beyond this one drug.
The Incretin System: GIP and GLP-1 in Brief
To understand what tirzepatide does, it helps to first understand the two hormones it imitates. GIP and GLP-1 are both members of the glucagon/secretin peptide superfamily, and both act through class B G-protein-coupled receptors—a family characterized by a large extracellular domain that captures the peptide’s C-terminus and a transmembrane bundle that the peptide’s N-terminus inserts into to trigger signaling.2 Despite this shared architecture, the two hormones have distinct physiology, and that distinction is central to the tirzepatide story.
GLP-1, secreted by intestinal L-cells after eating, has a broad and well-mapped set of actions. It enhances glucose-stimulated insulin secretion from pancreatic beta cells, suppresses glucagon release from alpha cells when glucose is elevated, slows gastric emptying through vagal pathways, and engages satiety circuits in the hypothalamus and brainstem to reduce food intake.1,6 Critically, GLP-1’s glucose-lowering effects are largely preserved in type 2 diabetes, which is why GLP-1 receptor agonists became a therapeutic mainstay.
GIP, secreted by K-cells, is also a potent stimulator of meal-time insulin secretion in healthy physiology.6 Its effects on the pancreatic alpha cell are more nuanced: GIP can promote glucagon secretion under some conditions, a difference from GLP-1 that has practical consequences discussed later. GIP also acts on adipose tissue, where its role in lipid handling has fueled a long-running controversy about whether activating or blocking the GIP receptor is the better strategy for weight loss.11 In addition, GIP receptors are expressed in regions of the central nervous system involved in appetite and nausea, and emerging work suggests central GIP signaling contributes meaningfully to the metabolic phenotype of dual agonists.11
Both native incretins share a practical weakness as drugs: they are rapidly degraded by the enzyme dipeptidyl peptidase-4 (DPP-4), giving them half-lives of only a few minutes. Any incretin-based medicine must therefore solve the problem of durability, whether through DPP-4 inhibition, structural modification that resists the enzyme, or albumin-binding lipidation as in tirzepatide.7,9 Understanding these baseline pathways—what each hormone does and how quickly it disappears—is the prerequisite for appreciating how tirzepatide re-engineers the whole system.
A further subtlety concerns the state of the two systems in disease. In type 2 diabetes, the overall incretin effect is diminished, and a substantial body of physiology attributes much of that loss specifically to a blunted insulinotropic response to GIP, while the response to GLP-1 is comparatively better preserved.6 This observation was historically read as a reason to give up on GIP as a diabetes target: if the beta cell no longer responds well to GIP in the very patients who need help, why bother? Tirzepatide reframes that logic. Emerging thinking holds that the GIP resistance seen in poorly controlled diabetes may be at least partly reversible once glycemia improves, so that a dual agonist could restore GIP responsiveness as it lowers glucose, rather than being permanently handicapped by it.11 Whether this “resensitization” genuinely contributes to the drug’s effect in humans remains an open question, but it illustrates how the same physiological facts can support opposite therapeutic conclusions depending on assumptions.
The two receptors also differ in their tissue distribution beyond the pancreas, which matters because tirzepatide’s effects are not confined to insulin secretion. GLP-1 receptors are found in the pancreas, the gut, the vagal afferents, the hypothalamus and brainstem, the heart, and the kidney. GIP receptors are expressed in the pancreas, in adipose tissue, in bone, and in several central nervous system regions.11 This overlapping-but-distinct map means that co-activating both receptors casts a wider physiological net than activating either alone—part of the theoretical appeal of the twincretin strategy, and part of why its full set of consequences is still being characterized.
Molecular Mechanism: Dual, Imbalanced, and Biased Agonism
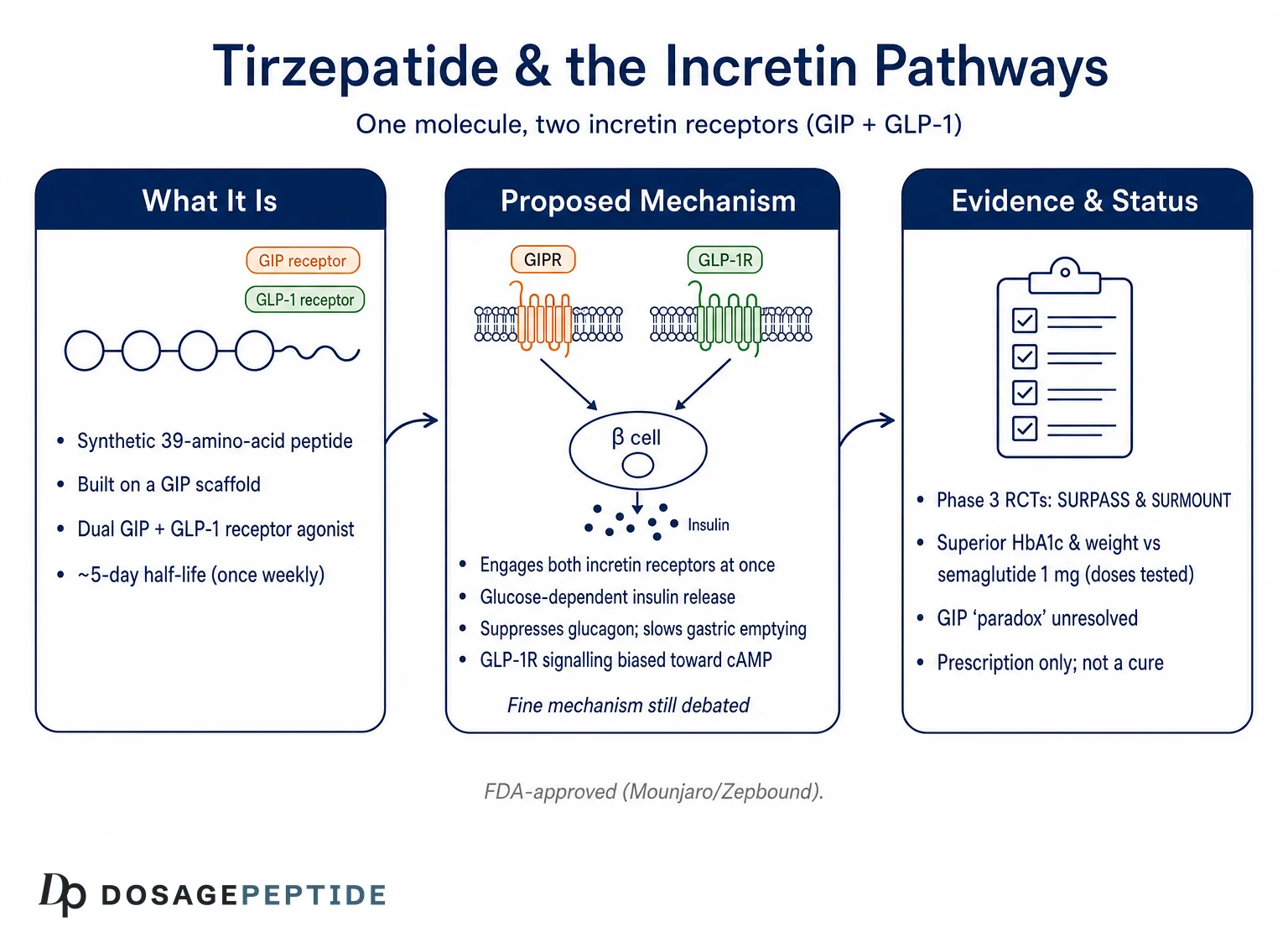
The most scientifically interesting feature of tirzepatide is not simply that it hits two receptors, but how it hits them. Detailed pharmacology studies describe it as an imbalanced and biased dual agonist, and both terms carry specific meaning.1
“Imbalanced” refers to differing affinity at the two receptors. Tirzepatide binds the GIP receptor with an affinity essentially equal to that of native GIP, but it binds the GLP-1 receptor with roughly five-fold weaker affinity than native GLP-1.1 In other words, at the molecular level the compound is more “GIP-like” than “GLP-1-like” in how tightly it engages each target. Structural studies using cryo-electron microscopy have mapped why: because the peptide is built on a GIP scaffold, its contacts with the GIP receptor closely mimic the native hormone, whereas its engagement of the GLP-1 receptor relies on high-affinity binding to the extracellular domain combined with a destabilizing influence from the lipid moiety.2
“Biased” refers to which downstream signals the receptor sends once activated. A class B GPCR can couple to at least two arms after binding: it can stimulate the Gs protein to generate the second messenger cyclic AMP (cAMP), and it can recruit β-arrestin, which typically drives receptor internalization and desensitization. Tirzepatide activates the GIP receptor in a manner that resembles native GIP, but at the GLP-1 receptor it is biased toward cAMP generation and away from β-arrestin recruitment, so it drives less receptor internalization than GLP-1 itself.1 The proposed functional consequence is reduced desensitization—the GLP-1 receptor stays responsive rather than being pulled inside the cell and switched off.
Why might that matter? In primary pancreatic islets, laboratory work found that β-arrestin1 normally limits the insulin response to GLP-1 but not to GIP or to tirzepatide, suggesting that tirzepatide’s biased signaling could actually enhance insulin secretion relative to what an unbiased GLP-1 ligand achieves.1 This is an elegant hypothesis, and it is supported by mechanistic data—but it remains a hypothesis about mechanism, not a proven explanation for the drug’s clinical superiority. Readers should hold the biased-agonism model as a leading and well-evidenced idea rather than settled fact. The table below summarizes the receptor-level comparison.
It is worth pausing on why receptor internalization and desensitization matter for a once-weekly drug. When a conventional agonist binds a GPCR, the cell frequently responds by recruiting β-arrestin, pulling the receptor inside the membrane, and either recycling or degrading it—a self-limiting brake that prevents runaway signaling. For a chronically dosed medicine that maintains steady drug levels for days, aggressive desensitization could blunt the very response the drug is meant to sustain. A ligand that generates strong second-messenger signaling while triggering comparatively little internalization could, in principle, keep the receptor at the cell surface and responsive over the long dosing interval. This is the mechanistic logic behind interest in tirzepatide’s GLP-1-receptor bias, and it connects the molecular pharmacology directly to the practical fact of weekly, rather than daily, administration.1,7
Two important caveats keep this from becoming an overclaim. First, signaling bias is notoriously assay-dependent and cell-context-dependent: the degree of bias measured in an engineered reporter cell line may not match what happens in a human beta cell, adipocyte, or neuron, so quantitative bias values should be read as directional rather than absolute.1 Second, bias at one receptor is only part of the story for a dual agonist whose net effect also depends on the balance between the two receptors, the local concentration of drug, and receptor reserve in each tissue. The honest summary is that tirzepatide’s distinctive signaling profile is real and well-characterized in vitro, plausibly relevant to its durability, and one of several factors—not a singular master switch—behind its clinical behavior.
| Property | Native GIP | Native GLP-1 | Tirzepatide |
|---|---|---|---|
| GIP receptor affinity | Reference (high) | Negligible | ~Equal to native GIP |
| GLP-1 receptor affinity | Negligible | Reference (high) | ~5-fold weaker than GLP-1 |
| GLP-1R signaling bias | — | Balanced | Favors cAMP over β-arrestin |
| GLP-1R internalization | — | Greater | Reduced vs GLP-1 |
| Plasma half-life | ~Minutes | ~Minutes | ~5 days |
Downstream Physiology: Insulin, Glucagon, Gut, and Brain
Receptor binding is only the first step. The physiologically important question is what dual incretin activation does to the organs that regulate glucose and body weight. Tirzepatide’s downstream effects broadly recapitulate the combined actions of GIP and GLP-1, though the relative contributions of each pathway are still being teased apart.
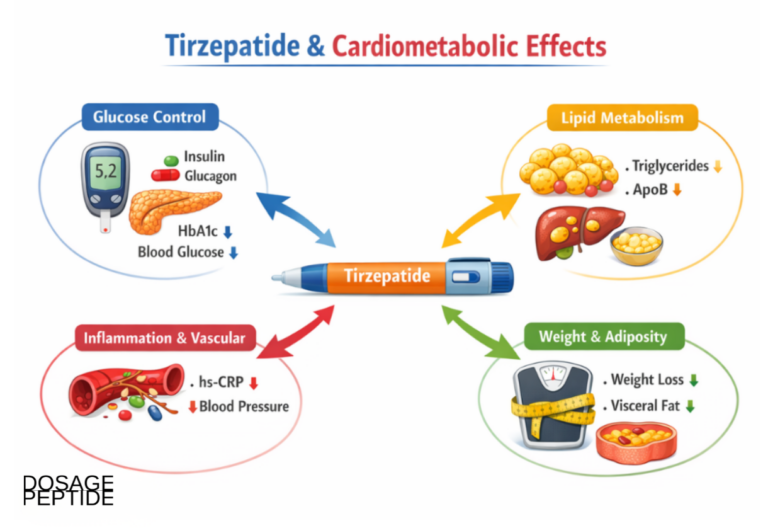
At the pancreatic beta cell, tirzepatide amplifies glucose-stimulated insulin secretion. Both incretin receptors contribute, and a notable feature is that the effect is glucose-dependent—insulin release is potentiated when blood glucose is elevated and tapers as glucose falls toward normal.7 This glucose dependence is why incretin-based therapies carry a low intrinsic risk of hypoglycemia when used without insulin or sulfonylureas. At the alpha cell, tirzepatide suppresses glucagon secretion in a glucose-dependent fashion, reducing hepatic glucose output during hyperglycemia; as glucose normalizes, both the insulin stimulus and the glucagon suppression ease off.7
Beyond the islet, tirzepatide slows gastric emptying, an effect largely attributed to the GLP-1 arm. This delay is most pronounced after the first dose and attenuates over time as tachyphylaxis develops, which is one reason nausea tends to be worst early in treatment and during dose escalation.7,12 Slowed gastric emptying blunts post-meal glucose excursions and contributes to satiety, but it also affects the absorption of co-administered oral drugs—a clinically relevant interaction.
The appetite and body-weight effects are where dual agonism becomes most intriguing. Reduced food intake is driven substantially by central action on hypothalamic and brainstem circuits that govern hunger and satiety. GLP-1 receptors in these regions are well characterized, but GIP receptors are also expressed centrally, and evidence increasingly implicates central GIP signaling in appetite regulation and possibly in mitigating GLP-1-associated nausea.11 In adipose tissue, GIP signaling influences lipid uptake, blood flow, and insulin sensitivity, though whether these peripheral fat effects meaningfully drive weight loss in humans is contested.11 Importantly, a study in human islets found that tirzepatide required a functional GIP receptor to elicit its full hormone-secretion response, providing direct evidence that the GIP arm is not merely decorative but functionally necessary for at least part of the drug’s action.3 Taken together, the physiology paints tirzepatide as a coordinated modulator of the entero-pancreatic-neural axis rather than a single-target agent. Our reconstitution and dosing reference tools exist for educational context only and do not alter this underlying biology.
Key Clinical Evidence and Its Honest Level
Unlike many compounds discussed in research circles, tirzepatide has a robust human evidence base, and it is worth stating the level of that evidence plainly. The pivotal data come from two large phase 3 randomized controlled trial programs—SURPASS in type 2 diabetes and SURMOUNT in obesity—published in leading peer-reviewed journals and reviewed by regulators.4,5 This is high-quality evidence by the usual hierarchy: prospective, randomized, double-blind, and adequately powered.
In SURPASS-2, a 40-week trial of 1,879 adults with type 2 diabetes on metformin, tirzepatide at 5, 10, and 15 mg produced greater reductions in HbA1c and body weight than semaglutide 1 mg, an established GLP-1 receptor agonist comparator.4 Across the broader SURPASS program, HbA1c reductions ranged roughly from 1.9 to 2.6 percentage points and body-weight reductions from about 6.6 to 13.9 percent depending on dose and background therapy.4 In SURMOUNT-1, a 72-week trial of 2,539 adults with obesity but without diabetes, mean weight reductions were approximately 16 percent at 5 mg, 21 percent at 10 mg, and 22 percent at 15 mg, versus about 2 to 3 percent with placebo.5 Roughly 90 percent or more of participants on active drug achieved at least 5 percent weight loss.
These are large effect sizes, and they justify genuine interest. But several caveats are essential for an honest reading. First, the comparison that made headlines—superiority over semaglutide 1 mg—was against a specific dose; higher semaglutide doses and newer agents change the competitive picture. Second, trial populations are selected and supported in ways that differ from everyday use; real-world adherence and results are typically more modest. Third, these trials measured surrogate and intermediate outcomes (HbA1c, weight, some cardiometabolic markers) over months to a few years. They do not establish that tirzepatide “treats” the long-term complications people ultimately care about, and weight regain after discontinuation has been documented, underscoring that the effect depends on continued exposure.
It is also worth distinguishing what the mechanism sections above can and cannot claim. The receptor pharmacology explains how the drug could produce these outcomes; it does not by itself prove that GIP agonism is why tirzepatide outperformed a GLP-1 monoagonist in a given trial, because trial design, dosing, and pharmacokinetics all contribute. Mechanistic plausibility and clinical superiority are related but not identical claims, and conflating them is a common error in popular summaries.
Comparisons and the GIP Agonism-versus-Antagonism Paradox
Placing tirzepatide alongside its relatives clarifies both its promise and the open questions. Compared with selective GLP-1 receptor agonists such as semaglutide, tirzepatide added the GIP arm and, in direct diabetes comparisons, achieved larger reductions in HbA1c and weight at the doses tested.4 Compared with the investigational triple agonist retatrutide—which adds glucagon-receptor activity—tirzepatide is the more mature, approved two-receptor agent. Readers comparing agents may find our semaglutide reference page and tirzepatide 15 mg reference useful for side-by-side context.
The deepest unresolved question in this field is what has been called the GIP paradox. For years, the dominant hypothesis held that blocking the GIP receptor should aid weight loss, because GIP promotes lipid storage in fat tissue, and because knocking out GIP receptors protects mice on a high-fat diet from obesity and insulin resistance.11 Yet tirzepatide activates the GIP receptor and produces substantial weight loss. Even more confusingly, GIP-receptor antagonists combined with GLP-1 agonism have also shown weight-loss benefit in early studies.11 How can both agonism and antagonism of the same receptor help?
Several explanations have been proposed, and none is fully proven. One argument notes that the “GIP drives weight gain” theory rests largely on loss-of-function knockout studies, with little direct evidence that GIP-receptor agonism increases adiposity; on this view, chronic agonism is genuinely beneficial through actions in the islet, brain, and fat.11 A competing hypothesis holds that chronic, high-level GIP-receptor agonism eventually causes desensitization—functionally silencing the receptor in a way that mimics antagonism.11 A third idea invokes a compensatory relationship between the two incretin receptors, whereby blunting one enhances signaling through the other. These competing models are actively debated in the literature, and a careful reader should treat the mechanism of GIP’s contribution as an open scientific question rather than a resolved textbook fact.11
The practical upshot is humility. Tirzepatide clearly works in trials, and the GIP arm is functionally required for its full effect in human islets.3 But the precise reason that adding GIP activity improves outcomes—and why the opposite manipulation can also help—is not yet settled, and future agents may exploit whichever hypothesis proves correct.
Beyond Glucose and Weight: Cardiometabolic and Pleiotropic Signals
Because incretin receptors are expressed in many tissues, tirzepatide’s influence extends beyond the two headline outcomes of blood sugar and body weight. Interpreting these broader signals requires care: some are well-supported secondary endpoints from randomized trials, others are exploratory findings or mechanistic inferences, and it would overstate the evidence to treat them all as established therapeutic benefits. With that caveat, several downstream effects are worth understanding mechanistically.
Blood pressure is one. Across the SURPASS program, tirzepatide was associated with modest reductions in systolic blood pressure, a pattern consistent with weight loss, natriuretic effects linked to GLP-1 receptor signaling in the kidney, and improved vascular function.4 Lipid profiles also tended to move favorably—reductions in triglycerides and adjustments in other fractions—again broadly attributable to the combination of weight loss and direct metabolic effects. These are plausible cardiometabolic benefits, but modest blood-pressure and lipid changes over months are intermediate markers, not proof of reduced cardiovascular events, which requires dedicated long-term outcome trials that are still maturing.
Body composition is a mechanistically interesting area. In SURMOUNT-1, weight loss reflected a roughly three-fold greater percentage reduction in fat mass than in lean mass, indicating that the drug preferentially reduces adipose tissue rather than muscle—though absolute lean-mass loss still occurs and is a topic of ongoing study, particularly in older adults where preserving muscle matters.5 The GIP arm is sometimes invoked here, given GIP’s roles in adipose biology, but attributing the favorable fat-to-lean ratio specifically to GIP versus GLP-1 versus the general physiology of caloric deficit is not something current human data can cleanly resolve.11
Other organ systems are under active investigation. Positive topline phase 3 results have been reported for tirzepatide in heart failure with preserved ejection fraction in the setting of obesity, and the SURMOUNT-OSA program supported an approval in obstructive sleep apnea—an effect largely mediated by weight loss reducing upper-airway and mechanical burden.13 Signals in metabolic dysfunction-associated steatotic liver disease and in chronic kidney disease are being explored, extending the incretin class’s reach. The disciplined reading is that these represent a widening frontier of investigation, some already validated by regulators and others still hypotheses, all flowing from the same broad tissue distribution of the two incretin receptors that tirzepatide engages.
Research Models and Methodology
The claims in this article rest on a layered methodology, and understanding those methods helps a reader weigh the evidence appropriately. The molecular characterization of tirzepatide—its affinities, its signaling bias, and its structural contacts—comes from in vitro pharmacology and structural biology. Radioligand and functional binding assays quantify how tightly the peptide engages each receptor; cAMP accumulation assays and β-arrestin recruitment assays (often using engineered cell lines) measure signaling bias; and cryo-electron microscopy resolves the atomic-level geometry of the peptide-receptor complexes.1,2 These techniques are powerful for mechanism but are performed in reductionist systems that may not perfectly predict whole-organism behavior.
Bridging toward physiology, researchers use isolated pancreatic islets—including human donor islets—to test whether receptor-level observations translate into hormone secretion. The finding that tirzepatide requires a functional GIP receptor for its full secretory effect in human islets came from exactly this kind of ex vivo model, strengthened by genetic and pharmacological tools to disable specific receptors.3 Rodent models, including receptor-knockout mice, have been indispensable for probing the roles of GIP and GLP-1 in adiposity and energy balance, and they are the source of much of the GIP-paradox debate—while also being a cautionary example of how species differences can mislead, since mouse fat biology does not map cleanly onto human clinical outcomes.11
At the top of the hierarchy sit the human pharmacokinetic and clinical studies. Population pharmacokinetic modeling, drawing on plasma concentrations from thousands of participants, established the roughly 5-day half-life, the near-80 percent subcutaneous bioavailability, the 99 percent albumin binding, and the roughly four-week approach to steady state that together justify once-weekly dosing.7,9 The phase 3 SURPASS and SURMOUNT trials, with randomized, double-blind, placebo- or active-controlled designs, provide the efficacy and safety data.4,5 Post-marketing pharmacovigilance, such as analyses of the FDA Adverse Event Reporting System (FAERS), adds real-world safety signal detection, albeit with the well-known limitations of spontaneous reporting—no denominator, reporting bias, and inability to prove causation.10
No single method is sufficient. The strength of the tirzepatide evidence base lies in its convergence: structural biology explains the binding, cell assays explain the signaling, islet studies confirm functional necessity of the GIP arm, pharmacokinetics explains the dosing, and randomized trials measure the outcomes. A weakness in any one layer—for instance, an unproven mechanistic hypothesis—does not undermine the layers above it, but it should temper confident storytelling about exactly why the drug works.
Safety and Tolerability
Any honest account of tirzepatide’s influence on incretin pathways must include the flip side of those same pathways: the adverse effects that flow directly from incretin biology. The dominant tolerability issue is gastrointestinal, a predictable consequence of GLP-1-mediated slowing of gastric emptying and central effects on nausea circuits.12 Across pooled trial data, gastrointestinal adverse events were dose-dependent, with pooled proportions of roughly 39 percent of participants at 5 mg, 46 percent at 10 mg, and 49 percent at 15 mg, with nausea and diarrhea most common.14 These events were most often mild to moderate, clustered around initiation and dose escalation, and tended to diminish over time—which is precisely why the approved titration schedule starts low and increases slowly.7 Discontinuation for adverse events was relatively uncommon and dose-related, generally in the single-digit percentage range across the studied doses.14
Concerns about pancreatitis have received close scrutiny given historical questions about incretin therapies. Pooled randomized-trial data have been reassuring: acute pancreatitis was rare and did not show a statistically significant excess over placebo in the obesity and diabetes programs, and the prescribing information reflects this by listing pancreatitis as a warning rather than a common event.5,8 Case reports of acute pancreatitis do exist, so the risk is not zero, but the controlled data do not support a large signal. Gallbladder-related events, including cholelithiasis, are recognized in the class and were reported slightly more often on drug than placebo in some analyses, though differences were generally modest.8
Because glucose-dependent insulin stimulation is intrinsic to incretin action, tirzepatide carries low hypoglycemia risk on its own, but that risk rises when it is combined with insulin or insulin secretagogues.7 The approved labeling also carries a boxed warning regarding thyroid C-cell tumors, based on rodent findings with the incretin class; the human relevance is uncertain, but it drives a contraindication in people with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2.8 Additional considerations documented in labeling and pharmacovigilance include the potential for dehydration-related acute kidney injury secondary to vomiting or diarrhea, and effects on the absorption of oral medications due to delayed gastric emptying.8,10
Several additional considerations round out the tolerability picture. Injection-site reactions and hypersensitivity have been reported but are generally uncommon and mild. As a large modified peptide, tirzepatide can in principle provoke anti-drug antibodies; in the trial program a proportion of participants developed such antibodies, but this did not translate into a clear loss of efficacy or a distinct safety signal in the aggregate data—an important reassurance, though immunogenicity is always monitored for peptide therapeutics.8 Delayed gastric emptying also creates a practical pharmacological interaction: because it can alter the rate and extent of absorption of co-administered oral drugs, particular attention is warranted for medications with narrow therapeutic windows and, per labeling, for oral contraceptive effectiveness around the time of initiation and dose increases.8
None of this is a substitute for individualized medical advice, and this article does not provide dosing recommendations. The point for a mechanistically minded reader is that tirzepatide’s benefits and its side effects arise from the same incretin pathways; one cannot fully separate the appetite-suppressing, glucose-lowering effects from the nausea and gut effects, because they share biological roots. This inseparability is why the slow titration schedule exists—it is an attempt to let the tolerability-limiting arms of incretin biology adapt while the therapeutic arms take hold, rather than a sign that the two can be pharmacologically decoupled.
Handling and Reconstitution in a Research Context
Approved tirzepatide products are supplied by the manufacturer as prefilled pens or single-dose vials formulated, stabilized, and quality-controlled for human use, and those are the only forms with an established safety and efficacy record. Some material discussed in research and educational settings, however, is supplied as a lyophilized (freeze-dried) powder intended for laboratory handling, and it is worth briefly addressing the physical chemistry involved—strictly as educational context, not as an endorsement of any non-clinical use.
As a peptide, tirzepatide is sensitive to the same degradation pathways that affect its native incretin cousins: hydrolysis, oxidation, aggregation, and physical denaturation from heat, agitation, or repeated freeze-thaw cycles. Lyophilized peptide is generally the most stable form and is typically stored cold and protected from light. Reconstitution—dissolving the powder into a liquid—is conventionally performed with bacteriostatic water (water containing a small percentage of benzyl alcohol as a preservative), added slowly down the vial wall rather than directly onto the peptide cake, followed by gentle swirling rather than vigorous shaking, because mechanical agitation can shear and denature peptide chains and promote foaming.7 The concentration that results depends on the mass of peptide and the volume of diluent, which is the arithmetic our educational reconstitution guide and 30 mg vial reference walk through for illustration.
From a mechanistic standpoint, handling matters because a degraded or aggregated peptide is not the same molecule that produced the receptor pharmacology described above. Aggregation can reduce the fraction of intact, correctly folded peptide available to bind the extracellular domains of the GIP and GLP-1 receptors, and oxidation of susceptible residues can alter binding. In other words, the elegant structural fit that underlies dual agonism depends on molecular integrity, and poor handling silently undermines it. This is one reason that pharmaceutical-grade manufacturing, with rigorous control of purity, endotoxin, and stability, is not a bureaucratic formality but a determinant of whether a preparation behaves as characterized.
It bears repeating that reconstitution arithmetic and handling chemistry are presented here only to explain peptide behavior. Nothing in this section should be read as guidance to prepare or administer tirzepatide outside of an approved, clinically supervised context, and the properly formulated commercial products remain the only versions with a validated clinical evidence base.
Limitations and the Human-Evidence Gap
Even for an approved drug with strong trial data, important limitations and unknowns remain, and naming them is part of an honest account. The first gap is mechanistic. As discussed, the biased-agonism model and the resolution of the GIP agonism-versus-antagonism paradox are not settled science; they are active, contested research areas.1,11 We know the GIP arm is functionally required for tirzepatide’s full effect in human islets, but we cannot yet fully quantify how much of the clinical benefit is attributable to GIP versus GLP-1 signaling in living humans, nor precisely by what tissue-level routes.3,11
The second gap concerns long-term and hard outcomes. The efficacy trials primarily measured surrogate and intermediate endpoints—HbA1c, body weight, blood pressure, lipids—over roughly one to three years.4,5 Dedicated cardiovascular outcome trials and other long-horizon studies are ongoing or maturing, but at the level of the general mechanistic literature it would be premature to claim that tirzepatide durably prevents the downstream complications of diabetes or obesity. Weight regain after stopping treatment also indicates that the intervention modifies physiology only while present, which shapes how its long-term value should be framed.
Third is the gap between trials and reality. Randomized trials enroll selected populations, provide structured support, and monitor adherence closely. Real-world effectiveness, tolerability, and persistence typically fall short of trial results, and adverse-event surveillance through spontaneous reporting systems like FAERS cannot establish causation or true incidence.10 Rare harms may emerge only with broad, prolonged use.
A fourth limitation, often overlooked, is measurement itself. Much of what we attribute to “incretin pathway” effects in humans is inferred indirectly, from circulating hormone levels, glucose and insulin dynamics, and imaging or body-composition surrogates, rather than from direct observation of receptor signaling in living human tissue. We cannot ethically biopsy a person’s pancreatic islets or hypothalamus during treatment to confirm that the biased-signaling and receptor-occupancy phenomena characterized in cells are occurring as modeled. This means the bridge from molecular mechanism to human physiology is built partly on plausible inference, and inference is where confident-sounding but under-evidenced claims tend to creep into popular accounts. Keeping that epistemic humility in view is part of reading this literature responsibly.
Finally, much of the provocative mechanistic biology—especially the case for or against GIP receptor targeting—derives from rodent and cell models whose translation to humans is imperfect.11 The history of GIP itself is a warning: a hormone once dismissed as therapeutically useless in type 2 diabetes turned out to be central to a breakthrough drug, which should make everyone cautious about confident extrapolation in either direction. For readers, the disciplined stance is to treat tirzepatide as a genuinely effective, well-studied, but incompletely understood medicine—strong where the randomized evidence is strong, appropriately uncertain where the mechanism and the long-term picture remain open.
Regulatory Status
Tirzepatide is a fully approved prescription medicine, not an investigational or research-only compound, and its regulatory record is unusually well documented. The U.S. Food and Drug Administration approved it under the brand name Mounjaro on 13 May 2022 as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.8 It was subsequently approved under the brand name Zepbound on 8 November 2023 for chronic weight management in adults with obesity, or overweight with at least one weight-related comorbidity.13 In December 2024, Zepbound also became the first medication approved specifically for moderate-to-severe obstructive sleep apnea in adults with obesity, supported by the SURMOUNT-OSA trial.13 Regulatory authorizations have followed in other jurisdictions, including the European Union.
These approvals are important context for interpreting the compound. They mean that tirzepatide’s manufacturing, purity, stability, labeling, and risk information have been reviewed by regulators, and that the approved products carry defined indications, contraindications, warnings—including the boxed warning for thyroid C-cell tumors—and titration schedules.8 It also means that legitimate use is as a prescribed therapeutic under medical supervision, dispensed as the manufacturer’s formulated pens or vials.
A regulatory caveat matters for the research-peptide audience specifically. Material sold as tirzepatide outside the pharmaceutical supply chain—often labeled “for research use only”—is not FDA-approved, is not manufactured to the same standards, and has not been evaluated for the purity, potency, sterility, or stability that the clinical evidence base assumes. The favorable trial results described in this article apply to the approved products studied in those trials, not to arbitrary preparations. New indications continue to be investigated—including heart failure with preserved ejection fraction in the context of obesity, where positive topline phase 3 results have been reported but which is not, as of this writing, an approved indication.13 As always, indication status and labeling evolve, and the regulator’s current documents are the authoritative source.
Frequently Asked Questions
Is tirzepatide FDA approved, or is it experimental?
Tirzepatide is FDA approved. It is marketed as Mounjaro for type 2 diabetes (approved May 2022) and as Zepbound for chronic weight management (approved November 2023) and for moderate-to-severe obstructive sleep apnea in adults with obesity (December 2024).8,13 It is not an experimental or research-only compound, although some material sold outside the pharmaceutical supply chain is not approved and has not undergone the same quality control.
What makes tirzepatide different from a GLP-1 drug like semaglutide?
Semaglutide is a selective GLP-1 receptor agonist. Tirzepatide activates two incretin receptors—GIP and GLP-1—from a single molecule built on a GIP-based scaffold.1,2 In the SURPASS-2 diabetes trial, tirzepatide produced greater HbA1c and weight reductions than semaglutide 1 mg at the doses tested, though the comparison was against a specific dose and does not settle every head-to-head question.4
Why is adding GIP activity beneficial if some research suggests blocking GIP also helps?
This is the genuine “GIP paradox,” and it is unresolved.11 Proposed explanations include the idea that chronic GIP agonism is directly beneficial in the islet, brain, and fat; that sustained agonism causes desensitization mimicking antagonism; or that manipulating one incretin receptor compensatorily changes signaling through the other. Human islet data confirm the GIP arm is functionally required for tirzepatide’s full effect, but the mechanism remains debated.3,11
What does “biased agonism” mean for tirzepatide?
At the GLP-1 receptor, tirzepatide favors cyclic-AMP signaling over β-arrestin recruitment, causing less receptor internalization than native GLP-1.1 The proposed consequence is reduced desensitization and potentially enhanced insulin secretion. This is a well-evidenced mechanistic hypothesis rather than a proven explanation for the drug’s clinical results.
How does tirzepatide affect blood sugar without causing frequent hypoglycemia?
Its stimulation of insulin and suppression of glucagon are glucose-dependent: they are strongest when blood glucose is high and taper as glucose approaches normal.7 This built-in glucose dependence gives incretin therapies a low intrinsic hypoglycemia risk—though that risk rises when combined with insulin or sulfonylureas.
What are the most common side effects, and why do they happen?
Gastrointestinal effects—nausea, diarrhea, vomiting, constipation—are most common and are dose-dependent, with pooled proportions of roughly 39 to 49 percent of trial participants depending on dose.14 They stem largely from GLP-1-mediated slowing of gastric emptying and central nausea circuits, are usually mild to moderate, and tend to ease over time, which is why dosing is titrated slowly.7,12
Does tirzepatide cure diabetes or obesity?
No. It modifies physiology to improve glycemic control and reduce body weight while it is being taken, but it does not cure these conditions.4,5 Weight regain after discontinuation has been documented, indicating that the effect depends on continued exposure rather than a permanent change.
Why does peptide handling and reconstitution matter mechanistically?
Tirzepatide’s dual agonism depends on an intact, correctly folded peptide that fits the extracellular domains of both receptors.2 Degradation from heat, agitation, oxidation, or repeated freeze-thaw can reduce the fraction of active molecule, silently undermining the pharmacology—one reason pharmaceutical-grade manufacturing and the approved formulations matter.
References
- Willard FS, Douros JD, Gabe MBN, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020;5(17):e140532. https://pmc.ncbi.nlm.nih.gov/articles/PMC7526454/
- Sun B, Willard FS, Feng D, et al. Structural determinants of dual incretin receptor agonism by tirzepatide. Proc Natl Acad Sci USA. 2022;119(13):e2116506119. https://pmc.ncbi.nlm.nih.gov/articles/PMC9060465/
- El K, Douros JD, Willard FS, et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nature Metabolism. 2023;5:945–954. https://www.nature.com/articles/s42255-023-00811-0
- Frías JP, Davies MJ, Rosenstock J, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes (SURPASS-2). N Engl J Med. 2021;385:503–515. https://www.nejm.org/doi/full/10.1056/NEJMoa2107519
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide once weekly for the treatment of obesity (SURMOUNT-1). N Engl J Med. 2022;387:205–216. https://www.nejm.org/doi/abs/10.1056/NEJMoa2206038
- Nauck MA, Müller TD. From the incretin concept and the discovery of GLP-1 to today’s diabetes therapy. Front Endocrinol. 2019;10:260. https://pmc.ncbi.nlm.nih.gov/articles/PMC6497767/
- StatPearls. Tirzepatide. NCBI Bookshelf NBK585056. https://www.ncbi.nlm.nih.gov/books/NBK585056/
- Zepbound (tirzepatide) Prescribing Information, Eli Lilly / U.S. FDA. https://pi.lilly.com/us/zepbound-uspi.pdf
- Furihata K, Mimura H, Urva S, et al. Population pharmacokinetics of the GIP/GLP-1 receptor agonist tirzepatide. CPT Pharmacometrics Syst Pharmacol. 2024. https://pmc.ncbi.nlm.nih.gov/articles/PMC10962491/
- Real-world safety profile of tirzepatide: pharmacovigilance analysis of the FDA Adverse Event Reporting System (FAERS) database. 2024. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11473560/
- A contemporary rationale for agonism of the GIP receptor in the treatment of obesity. Diabetes. 2025;74(8):1326. https://pmc.ncbi.nlm.nih.gov/articles/PMC12278790/
- Tirzepatide-induced gastrointestinal manifestations: a systematic review and meta-analysis. 2023. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10614464/
- Eli Lilly and Company. FDA approves Zepbound (tirzepatide) for moderate-to-severe obstructive sleep apnea in adults with obesity. Press release, 2024. https://investor.lilly.com/news-releases/news-release-details/fda-approves-zepboundr-tirzepatide-first-and-only-prescription
- Mishra R, Raj R, Elshimy G, et al. Adverse events related to tirzepatide: a systematic review and meta-analysis. J Endocr Soc. 2023;7(4):bvad016. https://pmc.ncbi.nlm.nih.gov/articles/PMC9915969/
Educational and research-informational content only. This article is not medical advice and does not provide dosing recommendations. Tirzepatide is an FDA-approved prescription medicine that should be used only under the supervision of a qualified healthcare professional; material sold outside the regulated pharmaceutical supply chain is not FDA-approved and has not been evaluated for safety, purity, or efficacy. Nothing here should be construed as encouraging any non-clinical preparation or use. Consult a licensed clinician for questions about diagnosis or treatment.