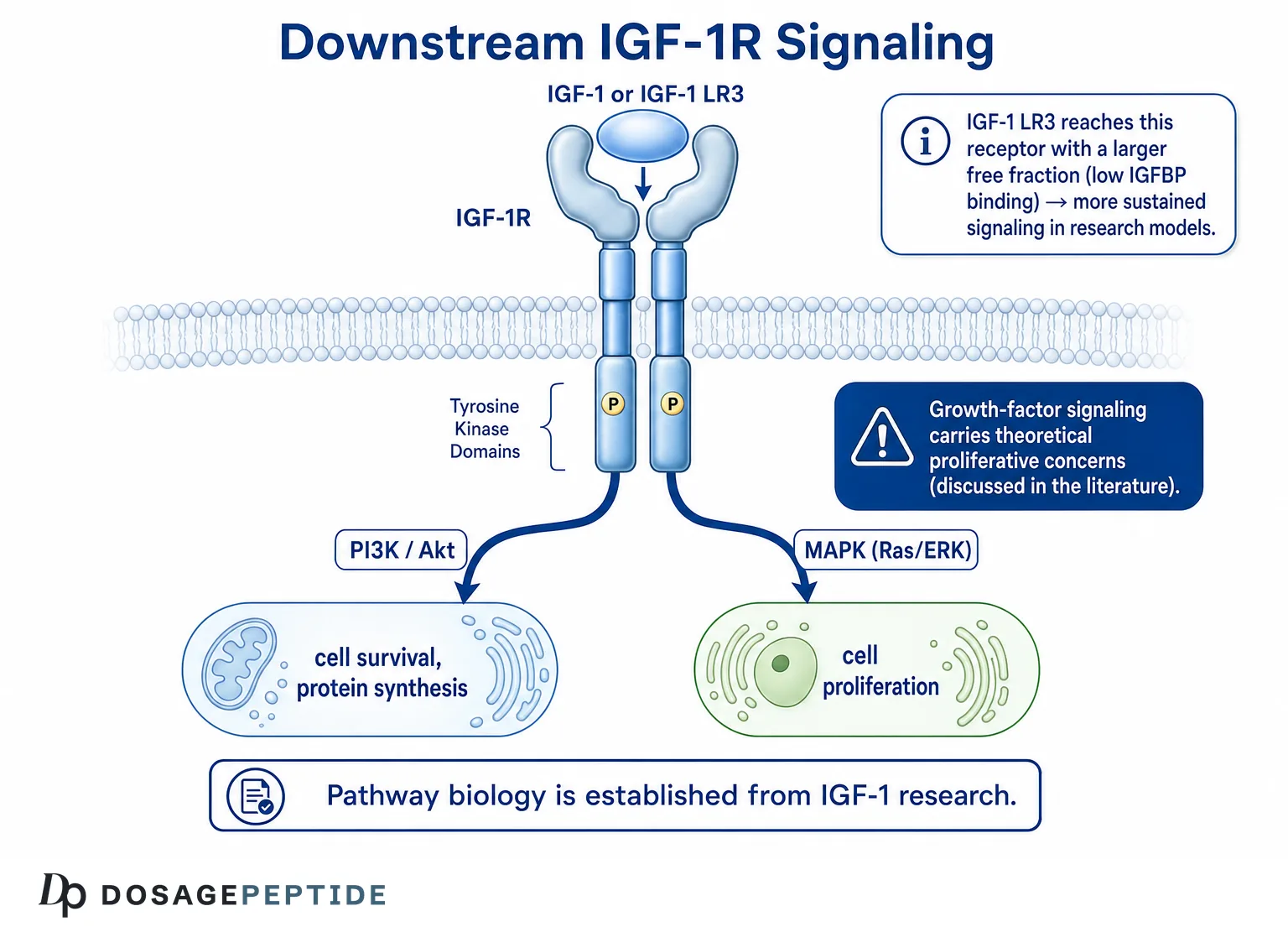
IGF-1 LR3 (Long R3 insulin-like growth factor-1) is an engineered analog of the naturally occurring peptide hormone IGF-1, redesigned in the laboratory so that it evades the binding proteins that normally sequester and regulate native IGF-1 in circulation. The central research question this article addresses is straightforward but frequently misunderstood: what exactly does the “Long R3” modification do at the molecular level, how does it alter the peptide’s half-life and receptor behavior, and how does IGF-1 LR3 differ from both native IGF-1 and the muscle-specific splice variant known as mechano-growth factor (MGF)? Because IGF-1 LR3 is a research reagent and not an approved human therapeutic, this reference material frames every claim by its evidence tier — separating what has been demonstrated in cell culture and animal models from what remains entirely untested in humans.
What Is the Research Context for IGF-1 LR3?
To understand IGF-1 LR3, it helps to first place native IGF-1 within the endocrine system. Insulin-like growth factor-1 is a 70-amino-acid single-chain polypeptide whose structure is homologous to proinsulin. It is produced predominantly in the liver in response to growth hormone (GH) stimulation, and it mediates a substantial fraction of GH’s anabolic and growth-promoting effects. IGF-1 was originally described as “sulfation factor” and later as “somatomedin C,” reflecting the historical recognition that GH does not act directly on many peripheral tissues but rather works through this intermediary.[1]
In the bloodstream, more than 95% of circulating IGF-1 is not free. It is bound to a family of six high-affinity insulin-like growth factor binding proteins (IGFBP-1 through IGFBP-6), with the great majority carried in a ternary complex composed of IGF-1, IGFBP-3, and an acid-labile subunit (ALS). This carrier system extends the circulating half-life of native IGF-1 from minutes to hours and tightly regulates how much free, bioactive peptide is available to engage receptors at any moment.[2]
IGF-1 LR3 was created specifically to circumvent this binding-protein control. It emerged from a body of protein-engineering work in the late 1980s and early 1990s — much of it from the Australian laboratories associated with Francis, Ballard, and Wallace — aimed at producing IGF analogs with reduced IGFBP affinity but preserved receptor activity, in order to study the relative contributions of binding-protein interaction versus receptor binding to biological potency.[3] The practical result of that research is a molecule that behaves very differently from native IGF-1 in laboratory systems, and that has since become a widely stocked reagent for cell-culture applications.
Why the distinction between reagent and therapeutic matters
This is the single most important framing point in the entire topic. Native IGF-1 exists as an FDA-approved human drug: recombinant human IGF-1, marketed as mecasermin (brand name Increlex), is approved for the treatment of growth failure in children with severe primary IGF-1 deficiency and in patients who have developed neutralizing antibodies to growth hormone.[4] IGF-1 LR3 is not mecasermin, is not the same molecule, and has never been approved by any regulatory agency for any human indication. It is sold and used as a laboratory research chemical — frequently as a serum-free cell-culture supplement — and human clinical trials evaluating IGF-1 LR3 as a therapeutic do not exist. Any discussion of IGF-1 LR3 research must keep that boundary explicit, and readers can explore the broader terminology in our peptide research glossary.
What Is IGF-1 LR3 at the Molecular Level?
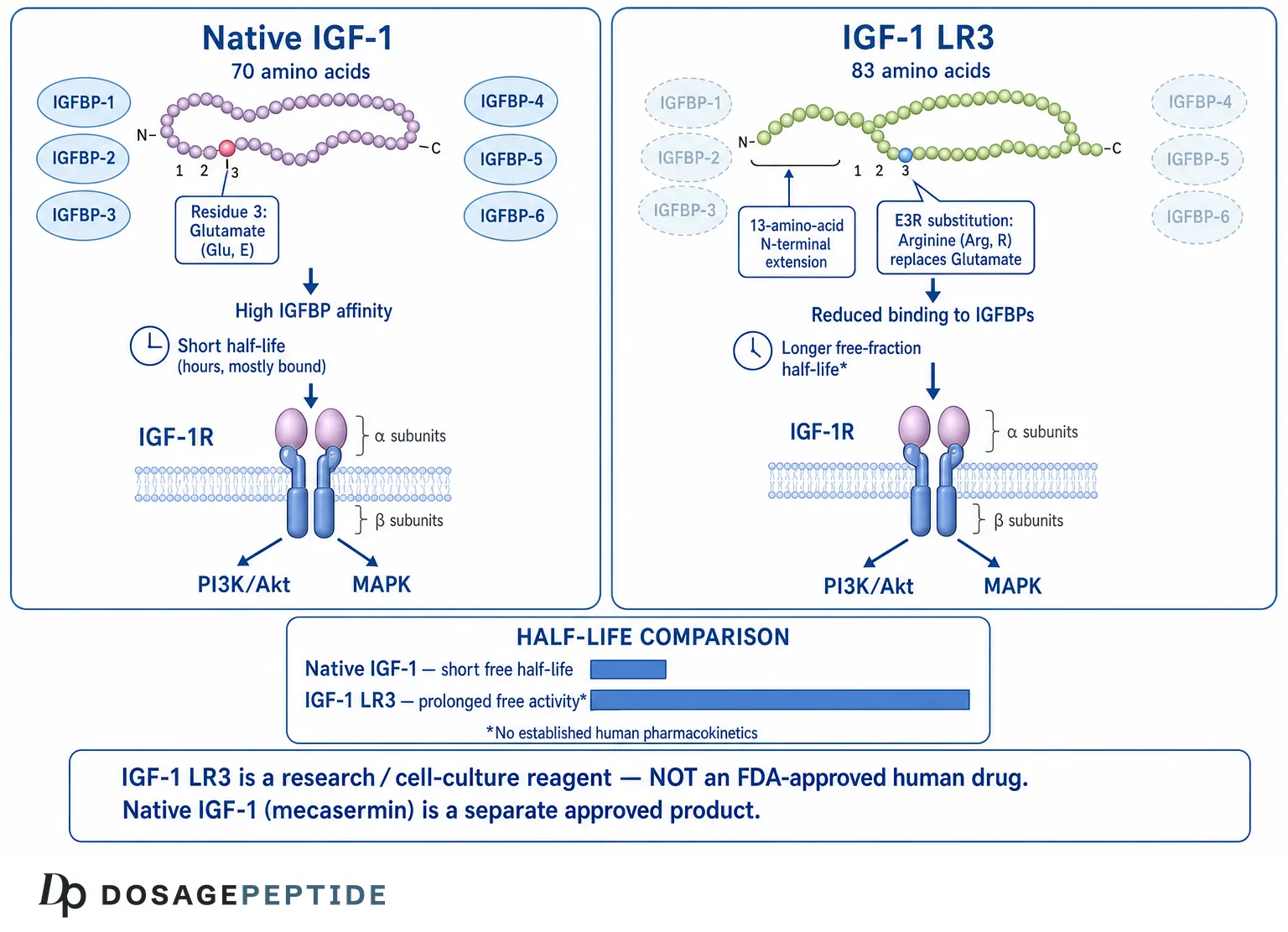
The name “Long R3 IGF-1” is a compact description of two distinct engineering changes made to the native IGF-1 sequence. Both changes are worth unpacking carefully, because together they explain essentially all of the reagent’s distinctive pharmacology.
The “R3” substitution
The “R3” refers to a single amino acid substitution at position 3 of the mature IGF-1 chain, where the native glutamate (Glu, E) residue is replaced with arginine (Arg, R). This region of the IGF-1 molecule sits within the portion of the peptide that contacts IGF binding proteins. Substituting arginine for glutamate at this position markedly reduces the analog’s affinity for the IGFBPs while leaving its ability to engage the type 1 IGF receptor (IGF-1R) largely intact.[3] In other words, the R3 change selectively degrades one interaction (binding-protein capture) without abolishing the other (receptor signaling).
The “Long” N-terminal extension
The “Long” prefix denotes a 13-amino-acid peptide extension fused to the N-terminus of the molecule. This extension is derived from a portion of a porcine growth hormone sequence and adds additional mass and structural context at the amino end of the chain. The N-terminal extension further reduces IGFBP binding and contributes to the analog’s enhanced stability and potency in the systems where it has been studied.[3] The combination of the extension plus the E3R substitution is what distinguishes “Long R3 IGF-1” from the simpler “R3 IGF-1” analog and from native IGF-1.
The net structural picture is a peptide of roughly 83 amino acids (the 70-residue IGF-1 core, modified at position 3, plus the 13-residue N-terminal extension), with a molecular weight of approximately 9.1–9.2 kDa — noticeably larger than native IGF-1’s ~7.6 kDa. These are the physical features that the downstream biology follows from. It is worth stressing that neither modification was intended to make the molecule “better” in any therapeutic sense; both were engineering choices made to answer a specific scientific question about how much of IGF-1’s potency is masked by binding-protein capture. The enhanced potency is a consequence of that experimental design, not evidence of suitability for any use in a living human being.
| Feature | Native IGF-1 | IGF-1 LR3 |
|---|---|---|
| Amino acid length | 70 residues | ~83 residues (70 + 13-residue N-terminal extension) |
| Residue at position 3 | Glutamate (Glu/E) | Arginine (Arg/R) |
| Approx. molecular weight | ~7.6 kDa | ~9.1–9.2 kDa |
| IGFBP affinity | High | Substantially reduced |
| IGF-1R engagement | High | Retained (receptor-active) |
| Regulatory status | FDA-approved as mecasermin (Increlex) | Research reagent only; no approval |
How Does IGFBP Binding Govern the Long R3 IGF-1 Half-Life?
The concept of IGFBP binding is the hinge on which the entire IGF-1 LR3 story turns, so it deserves a dedicated section. In native physiology, the six IGFBPs perform several overlapping regulatory jobs: they extend the circulating half-life of IGF-1, they create a reservoir of hormone that can be released as needed, they modulate the amount of free peptide available to receptors, and in some contexts they exert IGF-independent effects of their own.[2]
The six binding proteins and their distinct roles
The IGFBP family is not a single carrier but six related proteins with overlapping yet distinct functions, and appreciating their diversity clarifies exactly what IGF-1 LR3 was engineered to escape:
- IGFBP-1 is acutely regulated by insulin and nutritional state; its levels rise during fasting and fall after feeding, allowing it to modulate free IGF-1 availability in response to metabolic conditions.
- IGFBP-2 is the second most abundant binding protein in adult serum and is often elevated in catabolic states and in certain cancers, where it has attracted study as a biomarker.
- IGFBP-3 is the dominant carrier, responsible for the ternary complex with IGF-1 and the acid-labile subunit that accounts for the large, long-lived circulating IGF-1 pool. It is GH-dependent and also has IGF-independent pro-apoptotic activity.
- IGFBP-4 is generally inhibitory to IGF action and is regulated by the protease PAPP-A, which cleaves it to release bound IGF-1 in specific tissues.
- IGFBP-5 can be either stimulatory or inhibitory depending on context and binds to extracellular matrix, creating localized IGF reservoirs relevant to bone and muscle.
- IGFBP-6 preferentially binds IGF-2 over IGF-1 and is associated with growth-inhibitory effects.
These proteins collectively fine-tune IGF-1 signaling across tissues, time, and metabolic state. IGF-1 LR3’s reduced affinity applies broadly across this family, which is precisely why the analog behaves as if the entire regulatory layer has been dialed down.[2]
The paradox of binding proteins and half-life
There is an apparent paradox here that confuses many people first encountering the topic. For native IGF-1, binding to IGFBP-3 and ALS lengthens circulating half-life — the ternary complex protects the peptide from rapid renal clearance and proteolysis, so bound native IGF-1 persists in blood for hours rather than the roughly 10 minutes reported for free IGF-1. So how can reducing IGFBP binding, as the R3 and Long modifications do, be described as extending half-life?
The resolution is that the two situations describe different things. The binding proteins prolong the residence of native IGF-1 but, in doing so, keep the vast majority of it sequestered and biologically silent. An analog like IGF-1 LR3 that binds IGFBPs poorly does not join the large protected reservoir, but the fraction that is free and bioactive is dramatically higher, and — critically in laboratory and animal systems — that free analog is not being continually captured and buffered by binding proteins. In the anabolic-potency studies from Ballard and colleagues, IGF analogs that bind poorly to IGFBPs (including Long R3 IGF-1) were repeatedly more potent than native IGF-1 on a per-dose basis, precisely because more of the administered molecule remained available to receptors.[5]
What “longer half-life” means in practice
Estimates commonly cited for the biological half-life of IGF-1 LR3 — often quoted in the range of roughly 20–30 hours — should be read with real caution. These figures circulate widely in non-peer-reviewed reagent literature and hobbyist sources rather than in controlled human pharmacokinetic studies, which do not exist for this analog. What the peer-reviewed literature genuinely supports is more modest and more defensible: because IGF-1 LR3 escapes IGFBP capture, its free-fraction bioactivity is prolonged relative to native IGF-1 in the experimental systems studied, and its per-molecule anabolic potency is greater.[5] The precise numeric half-life in a human being is unknown, and any specific figure presented as established human pharmacokinetics is not evidence-based. Readers interested in how the IGF-1 axis is sustained through upstream signaling can compare this with our analysis of the molecular pathways linking tesamorelin stimulation to sustained IGF-1 elevation.
What Are the Mechanisms Studied for IGF-1 LR3 Signaling?
The mechanism of IGF-1 LR3 is, at the receptor level, the same mechanism as native IGF-1: both molecules act as agonists at the type 1 IGF receptor. Understanding how it works therefore means understanding IGF-1R signaling, which is one of the best-characterized growth-factor pathways in cell biology.
The type 1 IGF receptor (IGF-1R)
IGF-1R is a transmembrane receptor tyrosine kinase structurally related to the insulin receptor. It exists as a pre-formed heterotetramer of two extracellular alpha subunits and two transmembrane beta subunits linked by disulfide bonds. When IGF-1 (or an agonist analog such as IGF-1 LR3) binds the alpha subunits, the receptor undergoes a conformational change that activates the intrinsic tyrosine kinase domains of the beta subunits, triggering autophosphorylation and the recruitment of docking proteins.[6] Because IGF-1 LR3 retains strong IGF-1R affinity, it initiates the same receptor activation cascade — the difference lies in bioavailability, not in the fundamental signaling logic.
The PI3K/Akt pathway
The first major downstream branch is the phosphatidylinositol-3-kinase (PI3K)/Akt (protein kinase B) pathway. Activated IGF-1R phosphorylates insulin receptor substrate proteins (IRS-1 and IRS-2), which recruit and activate PI3K. PI3K generates the lipid second messenger PIP3, which activates Akt. Akt is the principal mediator of IGF-1’s pro-survival, anti-apoptotic, and anabolic effects: it phosphorylates and inhibits pro-apoptotic factors, activates the mechanistic target of rapamycin complex 1 (mTORC1) to drive protein synthesis, and suppresses the FOXO transcription factors that promote muscle atrophy and proteolysis.[7] In skeletal muscle models, this IGF-1–Akt–mTOR axis is a central driver of hypertrophic protein accretion and a brake on the atrophy program.
The Ras/MAPK pathway
The second major branch is the Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) cascade. Through adaptor proteins such as Shc and Grb2, activated IGF-1R engages the Ras GTPase, initiating the kinase relay that culminates in ERK1/2 activation. ERK signaling is broadly associated with cell-cycle progression, proliferation, and differentiation.[8] The balance between the PI3K/Akt (survival, metabolic, hypertrophic) arm and the MAPK (proliferative) arm is context-dependent, varying by cell type, receptor density, and the presence of co-receptors such as hybrid IGF-1R/insulin-receptor complexes.
Downstream effectors: mTOR, FOXO, and the atrophy–hypertrophy balance
Following the PI3K/Akt activation described above, several effectors determine the biological readout. Akt activates mTORC1, which phosphorylates the translational regulators S6K1 and 4E-BP1 to increase ribosomal biogenesis and cap-dependent translation — the molecular basis of increased protein synthesis. Simultaneously, Akt phosphorylates the FOXO family of transcription factors (FOXO1, FOXO3, FOXO4), sequestering them in the cytoplasm and preventing them from transcribing the “atrogenes” MuRF1 and atrogin-1/MAFbx that drive ubiquitin-proteasome-mediated muscle protein breakdown.[7] Through this dual action — promoting synthesis while suppressing degradation — IGF-1 signaling shifts the net protein balance toward accretion in the muscle models where it has been characterized.
Receptor crosstalk and specificity
IGF-1R does not operate in isolation. It shares approximately 60% sequence homology with the insulin receptor, and the two can form hybrid receptors composed of one IGF-1R half-receptor and one insulin-receptor half-receptor. These hybrids bind IGF-1 preferentially and add complexity to how IGF and insulin signals are integrated. At sufficiently high concentrations, IGF-1 and its analogs can also engage the insulin receptor directly, which underlies the recognized hypoglycemic (insulin-like) effects of IGF-1 exposure. The type 2 IGF receptor (IGF-2R / cation-independent mannose-6-phosphate receptor) does not signal in the classical sense but acts as a clearance sink for IGF-2 and, to a lesser extent, participates in ligand disposal. IGF-1 LR3’s agonism is directed principally at IGF-1R, but this receptor ecosystem is part of why growth-factor signaling is difficult to isolate cleanly in a whole organism.[6]
Why bioavailability changes the biology without changing the pathway
It is worth emphasizing that IGF-1 LR3 does not activate a novel or exotic pathway. Its distinctive laboratory behavior comes entirely from the fact that a larger free fraction reaches IGF-1R and is not buffered by binding proteins. This is precisely why IGF-1 LR3 is prized as a cell-culture supplement: in serum-free or serum-reduced media, where binding proteins secreted by cells could otherwise dampen native IGF-1 activity, the LR3 analog delivers more consistent, sustained receptor stimulation at lower molar concentrations. The signaling logic is conserved; only the amount of ligand reaching the receptor differs.
How Is IGF-1 LR3 Produced and Characterized in Research?
Because IGF-1 LR3 is fundamentally a research reagent, understanding how it is manufactured and quality-controlled is part of understanding what the molecule actually is — and where uncertainty enters when preparations are of unknown provenance.
Recombinant expression
IGF-1 LR3 is produced by recombinant DNA technology, most commonly by expressing the engineered gene sequence in Escherichia coli. The bacterial host synthesizes the peptide chain, which must then be recovered, refolded to establish the correct disulfide bond pattern, and purified. IGF-1 and its analogs contain three intramolecular disulfide bonds that are essential for the correct three-dimensional fold and therefore for receptor binding; misfolded or scrambled-disulfide species are biologically inactive or aberrant. Correct refolding is one of the more technically demanding steps in producing a functional preparation.[3]
Purification and identity confirmation
After refolding, research-grade preparations are purified by chromatographic methods such as reverse-phase and ion-exchange high-performance liquid chromatography (HPLC). Identity and purity are confirmed with techniques including mass spectrometry (to verify molecular weight and thereby the correct sequence and modifications), analytical HPLC (to assess purity percentage), and bioassays measuring receptor activation or cell proliferation. Endotoxin testing is relevant for cell-culture use because bacterial lipopolysaccharide contamination can confound experiments and harm sensitive cell lines.
Why provenance matters for interpreting any data
These characterization steps are routine for reputable biological-reagent suppliers serving the research market. They are not guaranteed for material obtained through informal or unregulated channels. A preparation labeled “IGF-1 LR3” without accompanying certificates of analysis, mass-spectrometric identity confirmation, purity data, and endotoxin results cannot be assumed to be correctly folded, correctly sequenced, or accurately concentrated. This is a recurring theme in research-chemical quality: the label is not the molecule, and conclusions drawn from an uncharacterized preparation carry a large asterisk regardless of how careful the downstream experiment is.
How Does IGF-1 LR3 Compare With Native IGF-1?
Placing IGF-1 LR3 vs IGF-1 side by side clarifies both what the engineering achieved and where the two molecules genuinely diverge. The comparison is not merely academic — conflating the two is the source of most misinformation about the reagent.
| Dimension | Native IGF-1 (e.g., mecasermin) | IGF-1 LR3 |
|---|---|---|
| Sequence | 70-residue endogenous peptide | Engineered analog: E3R + 13-residue N-terminal extension |
| IGFBP interaction | Binds all six IGFBPs with high affinity | Markedly reduced IGFBP affinity |
| Free bioactive fraction | Low (<5% free in circulation) | High (largely unbuffered in study systems) |
| Relative per-dose potency (animal/cell) | Reference standard | Higher potency reported in multiple models |
| Receptor target | IGF-1R (also insulin receptor at high concentrations) | IGF-1R (retained agonist activity) |
| Human data | Extensive; FDA-approved indication | None as a therapeutic; reagent-only |
| Regulatory status | Approved drug (mecasermin/Increlex) | Research chemical; not approved anywhere |
The key functional difference: escape from regulation
Native IGF-1 is embedded in a sophisticated regulatory apparatus. Its bioavailability is titrated moment-to-moment by the abundance of IGFBP-1 (which fluctuates with insulin and nutritional state), by IGFBP-3 and ALS levels (which are GH-dependent), and by IGFBP proteases that release bound IGF-1 in specific tissues. IGF-1 LR3 was engineered to opt out of that regulation. From a research standpoint this is a feature — it produces cleaner, more reproducible receptor activation for studying downstream signaling. From a safety standpoint it is also the crux of concern: bypassing the physiological brakes on IGF signaling removes a layer of control that evolved for a reason.
What the potency comparisons actually showed
In the classic anabolic studies, IGF analogs with reduced IGFBP binding — including Long R3 IGF-1 — produced greater nitrogen retention, greater gut and organ growth, and more pronounced anabolic responses than equimolar native IGF-1 in rodents, particularly in catabolic states such as dexamethasone treatment or dietary restriction.[5] These are rat and cell-culture findings. They establish that the analog is biologically more potent per molecule; they say nothing about safety, efficacy, or appropriate use in humans, for which there is no evidence base.
Where Does IGF-1 LR3 Sit Downstream of GH and GH Secretagogues?
IGF-1 occupies a defined position in the somatotropic (GH/IGF-1) axis, and understanding that hierarchy clarifies why IGF-1 LR3 is conceptually different from the many peptides that act upstream of it.
The GH–IGF-1 axis
The axis runs from the hypothalamus to peripheral tissue in a cascade. The hypothalamus releases growth hormone-releasing hormone (GHRH) and, oppositely, somatostatin, which respectively stimulate and inhibit the pituitary. The anterior pituitary secretes growth hormone in pulses. GH then acts on the liver and other tissues to induce IGF-1 production. Circulating and locally produced IGF-1 mediates many of GH’s downstream effects and also feeds back negatively on both the hypothalamus and pituitary to restrain further GH release.[1]
Secretagogues act upstream; IGF-1 LR3 acts at the endpoint
A large family of research peptides — GHRH analogs such as tesamorelin, and growth-hormone secretagogues/ghrelin-receptor agonists — work by increasing endogenous GH pulses, which in turn raise the body’s own IGF-1 through the intact, self-regulating cascade. This is a fundamentally different intervention point from IGF-1 LR3. A secretagogue nudges an upstream node and lets physiology set the resulting IGF-1 level within feedback limits; IGF-1 LR3 is an exogenous receptor agonist applied at the very endpoint of the axis, bypassing the feedback loop entirely. Our discussion of the pathways linking tesamorelin to sustained IGF-1 elevation examines the upstream, secretagogue-driven side of this same axis in detail, and the contrast highlights why the two categories carry different mechanistic profiles.
This positioning also explains a common point of confusion: raising GH does not simply equate to administering IGF-1 LR3. The former operates through negative feedback and produces native, binding-protein-regulated IGF-1; the latter delivers an unregulated analog directly to receptors. The two are not interchangeable in mechanism, even though both touch the IGF-1 signaling node.
The feedback dimension has a further consequence worth noting. Because native IGF-1 suppresses GH release when it rises, the endogenous axis is self-limiting — the system resists runaway signaling. An exogenous IGF-1R agonist introduced at the endpoint is not sensed by that feedback loop in the same way and does not switch itself off through this mechanism; it also does not raise the body’s own regulated IGF-1 the way an upstream secretagogue does. This is a qualitative, mechanistic distinction between the two intervention categories, and it is why the research literature treats endpoint agonism and upstream secretagogue stimulation as fundamentally different manipulations of the somatotropic axis rather than two routes to the same outcome.
How Does IGF-1 LR3 Differ From MGF (IGF-1Ec)?
Among the most frequent points of confusion is the relationship between IGF-1 LR3 and mechano-growth factor (MGF). They are related at the level of the IGF-1 gene but are otherwise distinct entities, and the difference is instructive.
MGF is a natural splice variant; IGF-1 LR3 is a synthetic analog
The IGF-1 gene can be alternatively spliced to produce several isoforms that share the mature IGF-1 core but differ in their C-terminal “E-peptide” extensions. The isoform designated IGF-1Ec in humans (and originally described in stretched/damaged rodent muscle) was named mechano-growth factor because its expression is upregulated by mechanical loading and muscle damage.[9] MGF is thus a naturally occurring, mechanically responsive splice product of the same gene that yields IGF-1. IGF-1 LR3, by contrast, is not a natural isoform at all — it is a deliberately engineered analog created in the laboratory by mutation and fusion.
Different structural regions, different proposed roles
The two molecules also differ in where they are modified. IGF-1 LR3 is altered at the N-terminus (the 13-residue extension) and at position 3 (the E→R substitution), changes aimed at the IGFBP-binding region. MGF’s distinguishing feature is its unique C-terminal E-peptide (the Ec extension), which has been studied for potential IGF-1R-independent actions on muscle satellite cell activation and proliferation, separate from the mature IGF-1 domain’s receptor signaling.[10] In the research literature, MGF has been proposed to act early after muscle damage to mobilize satellite cells, whereas the systemic IGF-1 isoform (IGF-1Ea) and analogs like LR3 are framed as driving the later proliferative and anabolic phases — though these distinctions come largely from cell and animal work and remain incompletely resolved.
For readers who want the full treatment of the mechano-responsive isoform, our dedicated explainer on what MGF (mechano-growth factor) is and the corresponding MGF research reference page cover its biology in depth. The essential takeaway is that MGF and IGF-1 LR3 are cousins from the same gene family that were shaped by entirely different processes — natural alternative splicing versus deliberate protein engineering — and they are studied for different proposed roles.
| Attribute | IGF-1 LR3 | MGF (IGF-1Ec) |
|---|---|---|
| Origin | Synthetic engineered analog | Natural alternative splice variant of the IGF-1 gene |
| Structural modification | N-terminal 13-residue extension + E3R substitution | Unique C-terminal Ec E-peptide |
| Trigger for relevance | Designed to evade IGFBPs | Upregulated by mechanical load / muscle damage |
| Proposed primary study role | Sustained IGF-1R agonism in culture/animal models | Early satellite-cell activation (partly receptor-independent) |
| Human therapeutic status | None (reagent) | None (research peptide) |
What Is Known About Satellite Cells and Anabolic Biology?
Much of the interest in IGF-1 signaling for muscle stems from a genuinely robust body of preclinical science on IGF-1’s role in skeletal muscle growth and repair. It is important to attribute these findings to IGF-1 biology broadly — not to IGF-1 LR3 specifically, which was rarely the molecule tested in the landmark studies.
Satellite cells and the repair cycle
Skeletal muscle harbors a population of resident stem cells called satellite cells, which lie quiescent beneath the basal lamina until activated by loading, injury, or growth-factor signals. Upon activation they proliferate, differentiate into myoblasts, and fuse either to form new fibers or to donate nuclei to existing fibers, supporting hypertrophy and regeneration. IGF-1 is one of the best-studied positive regulators of this cycle, promoting satellite-cell proliferation and differentiation and driving protein synthesis in mature fibers through the Akt/mTOR pathway.[11]
The transgenic and gene-transfer evidence
Two landmark rodent studies anchor this literature. In one, viral-mediated overexpression of IGF-1 locally in skeletal muscle blocked the age-related decline in muscle mass and strength in mice.[11] In another, transgenic mice engineered to express a locally acting IGF-1 isoform (mIGF-1) in muscle sustained hypertrophy and preserved regenerative capacity into old age.[12] These experiments established, convincingly and in living animals, that locally elevated IGF-1 activity supports muscle maintenance and repair. What they did not do is test systemically injected IGF-1 LR3 in humans, and the leap from “local transgene in a mouse” to “injected analog in a person” is scientifically enormous and unsupported.
Crosstalk with myostatin and the growth-regulatory network
IGF-1 signaling does not act on a blank slate; it is embedded in a network of opposing and cooperating factors. Myostatin (GDF-8), a member of the TGF-beta superfamily, is a negative regulator of muscle mass that signals through SMAD transcription factors and tends to oppose the Akt/mTOR-driven hypertrophic program that IGF-1 promotes. The balance between IGF-1’s anabolic push and myostatin’s restraint is one axis of muscle-size regulation studied in animal models.[14] Local IGF-1 isoforms, mechanical loading, satellite-cell dynamics, and myostatin signaling together form the regulatory web within which any exogenous IGF-1 agonist would be acting — another reason that predicting the whole-organism effect of an isolated analog from single-pathway experiments is unreliable.
Distinguishing local from systemic IGF-1 action
A recurring theme in the muscle literature is that locally produced IGF-1 — generated within the muscle in response to loading or expressed from a transgene — appears especially effective at supporting hypertrophy and repair, potentially through autocrine and paracrine action on nearby satellite cells and fibers.[12] This is mechanistically different from delivering a systemic analog to the whole body. The local-versus-systemic distinction is one more reason the strong transgenic findings cannot be assumed to predict what a circulating IGF-1 LR3 dose would do, and it further separates the natural, loading-responsive biology (closer to the MGF concept) from the pharmacology of an engineered systemic agonist.
Relevance to tissue-repair research more broadly
IGF-1 signaling intersects with the wider field of musculoskeletal repair research, where a range of peptides are studied for their proposed effects on connective tissue, tendon, and muscle. Readers interested in how the evidence is weighed for another widely discussed repair-associated compound may find our review of the evidence supporting BPC-157 for musculoskeletal healing a useful companion, particularly as a model of how to separate strong preclinical signals from the absence of human confirmation — the same discipline this IGF-1 LR3 discussion demands.
What Is the Current Evidence Level for IGF-1 LR3?
This section states the evidence tier as plainly as possible, because it is where honest reference material most differs from marketing copy.
Tier-by-tier breakdown
- In-vitro / cell culture (established): IGF-1 LR3 is genuinely well-validated as a cell-culture reagent. It is used as a potent, IGFBP-resistant supplement to support cell growth, survival, and productivity in serum-free and serum-reduced media, including in bioprocessing and stem-cell applications. This is the strongest and most legitimate evidence category for the molecule.[3]
- Animal (preclinical, limited to potency/anabolism studies): Rodent studies from the 1990s demonstrated that Long R3 IGF-1 and related IGFBP-resistant analogs are more anabolically potent than native IGF-1 per dose.[5] These are proof-of-pharmacology studies, not safety or efficacy trials for any indication.
- Human clinical (absent): There are no controlled human clinical trials of IGF-1 LR3 as a therapeutic. It has not been evaluated for safety, dosing, efficacy, or long-term effects in people, and it is not registered as an investigational drug for such use. A search of the clinical-trials registry returns trials of native IGF-1/mecasermin, not of the LR3 analog.[13]
How the evidence for IGF-1 LR3 compares to native IGF-1
A useful calibration is to contrast the two evidence bases directly. Native IGF-1 (mecasermin) has been through the full regulatory pipeline: dose-ranging studies, controlled trials in the target population, characterized pharmacokinetics, a defined adverse-event profile, and ongoing post-marketing surveillance. That is what a genuine human evidence base looks like. IGF-1 LR3 has none of these — its record consists of biochemical characterization, in-vitro potency data, and rodent anabolism studies conducted for pharmacological insight rather than therapeutic development.[5] The two molecules are frequently conflated precisely because they act on the same receptor, but their evidence tiers could hardly be further apart. Recognizing that asymmetry is central to reading any IGF-1 LR3 research claim honestly, and it is the reason this reference repeatedly returns to the reagent-versus-drug distinction.
What can and cannot be claimed
From this evidence structure, the defensible statements are narrow: IGF-1 LR3 is a receptor-active, IGFBP-resistant IGF-1 analog that is more potent than native IGF-1 in cell and animal models and is a valuable culture reagent. The statements that are not supported by evidence are broad: any claim of proven human benefit, any specific human dose or half-life presented as established, any therapeutic indication, and any assertion that preclinical muscle findings translate to human outcomes. The gap between those two lists is the entire honest story. The IGF-1 LR3 research reference and handling page frames all quantitative details strictly as laboratory reference material for research settings, consistent with this evidence tier.
What Safety Signals and Oncologic Considerations Appear in the Literature?
Because IGF-1 LR3 is a growth-factor agonist that bypasses the body’s regulatory brakes on IGF signaling, the theoretical safety concerns discussed in the scientific literature deserve careful, non-alarmist attention. These are considerations drawn from IGF-1 biology generally; they are presented here as context for why unregulated IGF-1R agonism is treated cautiously in research, not as clinical findings about IGF-1 LR3 in humans (which do not exist).
Proliferation and cancer biology
IGF-1R signaling is mitogenic and anti-apoptotic — it promotes cell division and cell survival. These are the same properties that make it relevant to cancer biology. A substantial body of research links elevated IGF-1 activity and IGF-1R signaling to the proliferation and survival of many tumor types, and epidemiological studies have associated higher circulating IGF-1 with increased risk of certain cancers.[6] The concern with any unregulated IGF-1R agonist is precisely that it could stimulate the survival and proliferation of pre-neoplastic or neoplastic cells, which is why IGF-1R has been an oncology drug target for inhibition rather than activation.[8]
Approved-drug labeling as a reference point
The FDA-approved label for mecasermin (native IGF-1) provides the closest regulated analog and lists recognized risks of IGF-1 exposure, including hypoglycemia (owing to IGF-1’s insulin-like metabolic action), intracranial hypertension, tonsillar/adenoidal hypertrophy, and the requirement for caution regarding neoplasia; the label carries warnings and notes that mecasermin should not be used in the presence of active or suspected malignancy.[4] These are the documented risks of a regulated, native IGF-1 product administered under medical supervision. An unapproved, more-potent, IGFBP-resistant analog with no human safety data cannot be assumed to be safer, and by mechanism the same hazard categories are at least theoretically relevant. The IGFBPs that IGF-1 LR3 is designed to evade themselves have IGF-independent tumor-suppressive and pro-apoptotic roles in some tissues, so bypassing them removes another layer of physiological regulation.[2]
Metabolic considerations from insulin-like activity
Beyond proliferative concerns, IGF-1 shares structural and functional homology with insulin and can produce insulin-like metabolic effects, most notably a lowering of blood glucose. In the regulated setting of mecasermin therapy, hypoglycemia is a recognized and monitored adverse effect, managed in part by co-administration with food.[4] A more potent, binding-protein-resistant analog such as IGF-1 LR3 would, by mechanism, be expected to retain this hypoglycemic potential, and because it evades the buffering system that normally tempers free IGF-1 activity, the metabolic effect could in principle be less predictable. This is a mechanistic inference, not a human finding, but it illustrates why unregulated IGF-1R agonism is not treated casually in the scientific literature.
Tissue growth and structural effects
The mecasermin label also documents effects such as lymphoid tissue hypertrophy (including tonsillar and adenoidal enlargement) and reports of intracranial hypertension, reflecting IGF-1’s broad growth-promoting activity across tissues rather than a muscle-specific action.[4] These observations underscore that IGF-1 signaling is not selectively anabolic for skeletal muscle; the receptor is expressed widely, and systemic agonism engages many tissue types simultaneously. Any framing of IGF-1 LR3 as a targeted muscle agent is inconsistent with the pleiotropic biology of the IGF-1 receptor.
What Are the Limitations of the IGF-1 LR3 Evidence Base?
A candid limitations section is essential for a compound at this evidence tier. Several structural gaps constrain what anyone can honestly say about IGF-1 LR3.
Extrapolation gaps
The most consequential limitation is the chain of extrapolation required to connect the existing evidence to any human-relevant conclusion. The anabolic data come from rodents, often in artificial catabolic states, using administration routes and durations designed to probe pharmacology rather than model therapeutic use.[5] The muscle-repair data come largely from local gene transfer and transgenic overexpression of native IGF-1 isoforms — not from systemically administered LR3 analog.[12] Each link in the chain — species, molecule, route, and endpoint — introduces uncertainty, and they compound.
Pharmacokinetic uncertainty
Widely repeated figures for IGF-1 LR3’s half-life, potency multiples, and tissue distribution in humans are not backed by controlled human pharmacokinetic studies. The frequently cited “20–30 hour half-life” and “three times more potent” claims trace to reagent marketing and informal sources rather than peer-reviewed human data. Honest reference material should treat these as unverified.
Reagent quality and identity
Because IGF-1 LR3 is sold as a research chemical rather than a pharmaceutical, material obtained outside of validated laboratory supply chains is not subject to pharmaceutical-grade identity, purity, sterility, or potency controls. Peptide identity, correct disulfide folding, endotoxin content, and actual concentration can vary, which further undermines any attempt to reason about effects in a living organism from an uncharacterized preparation.
Publication age and reagent-literature bias
Much of the foundational, peer-reviewed pharmacology of Long R3 IGF-1 dates from the late 1980s and 1990s, when the analog was developed as a tool for dissecting IGFBP-versus-receptor contributions to potency and for biotechnology applications such as cell-culture media.[3] The scientific interest largely resolved the original questions and moved on. As a result, the modern information environment around IGF-1 LR3 is dominated not by fresh clinical science but by commercial reagent descriptions and non-peer-reviewed community sources, which is where inflated or unsourced numeric claims tend to originate. Readers should weight peer-reviewed primary literature far above such secondary material.
Absence of long-term and human safety data
Finally, and most simply: there are no long-term studies, no human trials, and no post-market surveillance for IGF-1 LR3, because it is not a human product. The theoretical oncologic and metabolic concerns that follow from IGF-1R agonism have never been assessed for this specific analog in people. The absence of evidence of harm is not evidence of absence of harm — it is the direct consequence of the compound never having been studied for human use. The research glossary defines the evidence-tier terminology used throughout this reference so that these distinctions remain unambiguous.
Frequently Asked Questions
Is IGF-1 LR3 the same thing as IGF-1?
No. IGF-1 LR3 is an engineered analog of native IGF-1, not the natural hormone. It carries a 13-amino-acid N-terminal extension (“Long”) and an arginine-for-glutamate substitution at position 3 (“R3”). These changes were designed to reduce its binding to IGF binding proteins while preserving its ability to activate the IGF-1 receptor. Native IGF-1 is regulated by binding proteins; IGF-1 LR3 was specifically built to escape that regulation, giving it different pharmacology in laboratory systems.
Why does IGF-1 LR3 have a longer half-life than native IGF-1?
The extended activity attributed to IGF-1 LR3 comes from its reduced affinity for IGF binding proteins. Native IGF-1 is largely captured by these proteins, which regulate how much free, active hormone is available. Because IGF-1 LR3 evades that capture, a larger fraction remains free and receptor-active in study systems. However, the widely quoted specific numbers (such as 20–30 hours) come from informal sources, not controlled human pharmacokinetic studies, which do not exist for this analog.
Is IGF-1 LR3 approved by the FDA?
No. IGF-1 LR3 is not approved by the FDA or any regulatory agency for any human use. It is sold and used as a laboratory research reagent, commonly as a cell-culture supplement. Native IGF-1 exists as an approved drug called mecasermin (Increlex) for severe primary IGF-1 deficiency, but mecasermin is a different molecule — it is native IGF-1, not the LR3 analog. There are no human clinical trials of IGF-1 LR3 as a therapeutic.
How does IGF-1 LR3 differ from MGF?
MGF (mechano-growth factor, the IGF-1Ec splice variant) is a natural alternative splice product of the IGF-1 gene, upregulated by mechanical loading and muscle damage, distinguished by a unique C-terminal E-peptide. IGF-1 LR3 is a synthetic analog modified at the N-terminus and position 3 to evade binding proteins. They come from the same gene family but arise by completely different processes — natural splicing versus deliberate protein engineering — and are studied for different proposed roles.
What signaling pathways does IGF-1 LR3 activate?
At the receptor level, IGF-1 LR3 activates the same pathways as native IGF-1 because it is an agonist at the type 1 IGF receptor (IGF-1R). Receptor activation triggers two major branches: the PI3K/Akt pathway, which drives survival, protein synthesis via mTOR, and suppression of atrophy signals; and the Ras/MAPK/ERK pathway, associated with proliferation and differentiation. The molecule does not activate a novel pathway — its distinctiveness lies in greater bioavailability, not in different signaling.
Does research on IGF-1 in muscle apply to IGF-1 LR3 in humans?
Not directly. The strong preclinical muscle findings — such as IGF-1 overexpression preventing age-related muscle loss in mice — used locally expressed native IGF-1 isoforms in animals, not systemically injected IGF-1 LR3 in people. Translating those results to humans would require crossing several large gaps: different species, different molecule, different delivery route, and different endpoints. No human studies bridge that gap, so the muscle-repair evidence should be attributed to IGF-1 biology broadly, not to IGF-1 LR3 specifically.
What are the theoretical safety concerns discussed for IGF-1 signaling?
The scientific literature discusses proliferative and oncologic concerns because IGF-1R signaling promotes cell growth and survival. Elevated IGF-1 activity has been associated with tumor cell proliferation and, epidemiologically, with certain cancer risks, which is why IGF-1R has been pursued as a target for inhibition in oncology. The approved native-IGF-1 label also notes hypoglycemia and intracranial hypertension. These are mechanism-based and native-drug considerations; no human safety data exist for the IGF-1 LR3 analog itself.
Why is IGF-1 LR3 used in cell culture?
In serum-free or serum-reduced culture media, cells may secrete binding proteins that dampen native IGF-1 activity, producing inconsistent stimulation. Because IGF-1 LR3 resists binding-protein capture, it delivers more sustained and reproducible IGF-1 receptor activation at lower concentrations, supporting cell growth, survival, and productivity. This is its best-validated and most legitimate application, spanning bioprocessing and stem-cell research, and it is the reason the analog was engineered in the first place.
Where does IGF-1 LR3 sit relative to growth hormone secretagogues?
Growth hormone secretagogues and GHRH analogs act upstream, increasing the body’s own GH pulses, which then raise endogenous IGF-1 through an intact, self-regulating feedback loop. IGF-1 LR3 acts at the opposite end of that axis, as an exogenous receptor agonist applied directly at the endpoint, bypassing the feedback control entirely. Raising GH is therefore mechanistically distinct from administering an IGF-1 analog, even though both ultimately touch IGF-1 receptor signaling.
References
- Laron Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol Pathol. 2001;54(5):311–316.
- Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. 2014;14(5):329–341.
- Francis GL, Ross M, Ballard FJ, et al. Novel recombinant fusion protein analogues of insulin-like growth factor (IGF)-I indicate the relative importance of IGF-binding protein and receptor binding for enhanced biological potency. J Mol Endocrinol. 1992;8(3):213–223.
- Increlex (mecasermin injection) Prescribing Information. U.S. Food and Drug Administration.
- Tomas FM, Lemmey AB, Read LC, Ballard FJ. Superior potency of infused IGF-I analogues which bind poorly to IGF-binding proteins is maintained when administered by injection. J Endocrinol. 1996;150(1):77–84.
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928.
- Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am. 2012;41(2):425–443.
- LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–137.
- Yang S, Alnaqeeb M, Simpson H, Goldspink G. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil. 1996;17(4):487–495.
- Hameed M, Orrell RW, Cobbold M, Goldspink G, Harridge SDR. Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J Physiol. 2003;547(Pt 1):247–254.
- Barton-Davis ER, Shoturma DI, Musarò A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci USA. 1998;95(26):15603–15607.
- Musarò A, McCullagh K, Paul A, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27(2):195–200.
- ClinicalTrials.gov registry search: mecasermin (native recombinant human IGF-1). U.S. National Library of Medicine.
- Philippou A, Maridaki M, Halapas A, Koutsilieris M. The role of the insulin-like growth factor 1 (IGF-1) in skeletal muscle physiology. In Vivo. 2007;21(1):45–54.
Research-use disclaimer: This article is educational reference material about IGF-1 LR3 as a laboratory research reagent. It is not medical advice, not a therapeutic recommendation, and not an endorsement of human use. IGF-1 LR3 is not approved by any regulatory agency for human administration, and no human clinical trials support such use. All handling and quantitative details are intended solely for controlled research settings by qualified personnel.