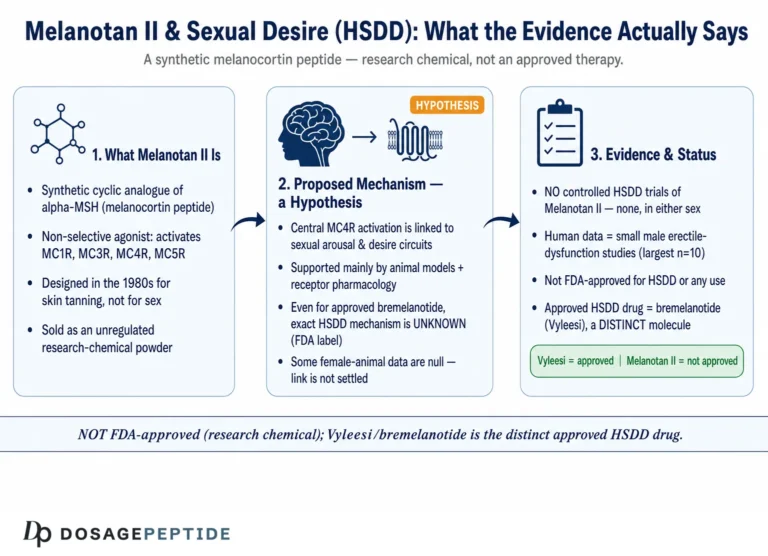
The question of how Melanotan II modulates melanocortin 1 receptor (MC1R) signaling sits at the intersection of receptor pharmacology and pigment biology. In controlled pigmentation research, Melanotan II is used as a metabolically stable, superpotent analog of alpha-melanocyte-stimulating hormone (α-MSH) to interrogate a single, well-mapped intracellular cascade: MC1R → Gs → adenylyl cyclase → cyclic AMP (cAMP) → protein kinase A (PKA) → CREB → MITF → tyrosinase and eumelanin synthesis. This article examines what that mechanism actually is, what the experimental evidence genuinely supports, and—critically—why the compound remains an unapproved research chemical carrying documented safety signals rather than an established therapeutic.
Melanotan II (MT-II) is a synthetic cyclic heptapeptide and a non-selective melanocortin receptor agonist. It engages MC1R (the pigmentation receptor) but also MC3R, MC4R, and MC5R, which is why appetite suppression and spontaneous erections appear alongside tanning in the descriptive literature. The properly approved, far better-characterized MC1R agonist is afamelanotide (Scenesse), a linear α-MSH analog approved for erythropoietic protoporphyria—a useful reference point for understanding what a rigorously studied melanocortin drug looks like. Everything below is framed strictly as pigmentation research and is not medical advice; Melanotan II is not approved for human use.
Why is MC1R the right receptor to study for pigmentation?
Before looking at Melanotan II specifically, it helps to establish why MC1R—out of the entire GPCR superfamily—is the pigmentation receptor worth probing. MC1R sits at the top of the melanocyte’s decision hierarchy. It is the gatekeeper that translates an extracellular hormonal cue (α-MSH released from keratinocytes, especially after UV exposure) into the intracellular program that determines both how much pigment a melanocyte makes and which kind. Genetic evidence makes the case forcefully: MC1R is the single strongest genetic determinant of human pigmentation phenotype, and its loss-of-function variants explain the red-hair, fair-skin, poor-tanning phenotype and its associated melanoma susceptibility. No other single locus concentrates as much of the pigmentation signal.
This is what makes MC1R a clean research target. Because the receptor is upstream of a linear, well-annotated cascade (cAMP → PKA → CREB → MITF → pigment enzymes), a stable agonist applied to it produces an interpretable chain of measurable events. Melanotan II is valuable precisely because it lets researchers push on this one node hard and reproducibly. The catch—returned to throughout this article—is that MT-II does not push on only that node, and MC1R itself varies from person to person.
What is Melanotan II and how does it relate to α-MSH?
Melanotan II descends directly from the endogenous melanocortin peptide α-MSH. The native hormone is a linear 13-residue peptide that activates MC1R on melanocytes, but it is degraded by serum peptidases within minutes, which makes it impractical as an experimental probe for sustained receptor engagement. In the 1980s, a University of Arizona group—Victor Hruby, Mac Hadley, Robert Dorr, and colleagues—set out to build a metabolically resistant, superpotent α-MSH analog, initially motivated by the idea that inducing melanin without ultraviolet (UV) exposure might reduce photodamage.[5]
The lineage produced two compounds relevant to pigmentation research. Melanotan I (afamelanotide) is a linear analog and is the molecule later developed and approved as Scenesse. Melanotan II is a cyclic peptide, structurally constrained by a lactam bridge, that trades receptor selectivity for extreme potency and stability. Two key modifications drive its behavior:
- Substitution of the labile methionine with norleucine (Nle4), removing an oxidation-prone residue and increasing stability.
- Substitution of L-phenylalanine with D-phenylalanine (D-Phe7) plus a lactam cyclization that locks the pharmacophore into the bioactive β-turn conformation, dramatically raising affinity and resistance to proteolysis.
The practical consequence for the laboratory is a ligand that binds MC1R with high affinity and stays intact long enough to produce a reproducible, sustained signaling readout—exactly what is needed to study receptor activation dynamics. The cost is selectivity: MT-II is effectively a pan-melanocortin agonist across MC1R, MC3R, MC4R, and MC5R (it does not meaningfully engage the adrenal MC2R). For a reference glossary of these receptor and pathway terms, see the Dosage Peptide peptide research glossary.
The melanocortin receptor family in one paragraph
Understanding MT-II research requires holding the whole melanocortin receptor family in view, because the compound touches four of the five members. MC1R is expressed on melanocytes and drives pigmentation. MC2R is the adrenal ACTH receptor and is not activated by MT-II—an important control point, since it means MT-II does not directly perturb cortisol biology. MC3R and MC4R are central-nervous-system receptors governing energy homeostasis, appetite, and sexual function; MC4R activation is the pharmacological basis for both MT-II’s appetite-suppressant and erectogenic effects. MC5R is involved in exocrine (e.g., sebaceous) gland function. Every pigmentation experiment with MT-II is therefore, unavoidably, also stimulating three non-pigmentary receptors—the single most important methodological caveat in the entire field.
How does the melanocortin system work?
Melanotan II is only interpretable against the endogenous system it imitates. That system begins not with a receptor but with a single precursor protein, proopiomelanocortin (POMC), and cascades through tissue-specific enzymatic processing to five receptors with distinct tissue roles. Mapping this system is what lets a researcher say precisely which parts of MT-II’s activity are “on-target” pigmentation and which are the unavoidable off-target melanocortin biology.
POMC and the birth of α-MSH
POMC is a large precursor polypeptide (a 241-residue protein produced after signal-peptide removal from pre-POMC) that carries, within its sequence, the messages for several distinct hormones. It is not itself active; it must be cut. Tissue-specific prohormone convertases (PC1/3 and PC2), together with the regulatory protein 7B2 and downstream carboxypeptidase and acetylation steps, cleave POMC at pairs of basic residues to liberate a family of peptides—ACTH, α-, β-, and γ-MSH, and β-endorphin among them. Which peptides emerge depends on which convertases a given cell expresses: the anterior pituitary favors ACTH, while the skin and hypothalamus complete the additional cuts that yield α-melanocyte-stimulating hormone (α-MSH), the 13-residue peptide that is MC1R’s principal endogenous ligand.[12]
Crucially for pigmentation, this machinery is present in the skin itself. Human epidermal keratinocytes and melanocytes synthesize and secrete POMC-derived peptides, and—this is the key regulatory link—ultraviolet B (UVB) radiation upregulates POMC transcription and the release of α-MSH and ACTH from these cells.[13] This is the molecular basis of the tan: UV damage in a keratinocyte prompts local α-MSH release, which acts in a paracrine fashion on the neighboring melanocyte’s MC1R to raise eumelanin. Melanotan II short-circuits this loop by supplying a stable, superpotent α-MSH surrogate directly, which is exactly why it produces pigmentation without the UV trigger—and why that decoupling is central to how the approved analog afamelanotide is used therapeutically.
The five melanocortin receptors and their tissue roles
The liberated melanocortin peptides act on a family of five class A GPCRs, all coupling primarily to Gs and cAMP but distributed across very different tissues. Their divergent roles are why a non-selective agonist like MT-II produces effects far beyond the skin:
| Receptor | Principal tissue | Physiological role | Engaged by MT-II? |
|---|---|---|---|
| MC1R | Melanocytes | Eumelanin synthesis, DNA-repair/antioxidant program; strongest genetic determinant of human pigmentation | Yes (the intended target) |
| MC2R | Adrenal cortex | ACTH receptor driving glucocorticoid synthesis; requires accessory protein MRAP | No (ACTH-selective) |
| MC3R | Hypothalamus, limbic system | Energy homeostasis, feeding, and circadian metabolic regulation | Yes (off-target) |
| MC4R | CNS (hypothalamus, brainstem) | Appetite/satiety and erectile function; basis of MT-II’s appetite and erectogenic effects | Yes (off-target) |
| MC5R | Exocrine glands (sebaceous, lacrimal, Harderian) | Sebum and exocrine-lipid production; knockout mice show defective sebaceous lipid output and water repulsion | Yes (off-target) |
The MC5R and MC4R roles are not speculative. Targeted disruption of MC5R in mice produces a defect in sebaceous-lipid production with impaired water repulsion and thermoregulation, establishing its exocrine role in vivo.[14] MC4R’s central role in feeding and erection is likewise well established and is the reason the MT-II lineage was successfully redirected into bremelanotide/PT-141. When a study reports that an MT-II dose caused nausea, appetite loss, or spontaneous erection, it is reporting MC3R/MC4R pharmacology, not an idiosyncratic side effect.
From receptor to pigment
The pigmentation output that MT-II drives is the terminal branch of the melanocortin system. It runs through the linear Gs–cAMP–PKA–CREB–MITF axis dissected step by step in the mechanism section below, with MITF then driving tyrosinase, TYRP1, and DCT to build eumelanin. That linearity is precisely what makes MC1R such a tractable research node: each experimental readout—from cAMP accumulation through spectrophotometric melanin content—maps cleanly onto one identifiable step of a single well-annotated cascade.
How does Melanotan II bind MC1R in pigmentation studies?
Melanotan II occupies the orthosteric binding pocket of MC1R—the same site the endogenous α-MSH core (His-Phe-Arg-Trp) engages. MC1R is a class A (rhodopsin-like) G-protein-coupled receptor (GPCR) with seven transmembrane helices, and the melanocortin pharmacophore inserts between these helices to stabilize the active receptor conformation. The lactam-cyclized backbone of MT-II pre-organizes the peptide into the receptor-competent geometry, so less binding energy is spent on conformational rearrangement, which is the structural basis for its high affinity.
Several features distinguish MT-II binding as a research probe:
- Cyclic constraint stabilizes the His-D-Phe-Arg-Trp message sequence in the bioactive conformation, improving both affinity and the reproducibility of receptor engagement across experimental replicates.
- D-Phe7 stereochemistry resists the peptidases that rapidly clear native α-MSH, extending the window of receptor occupancy in cell and tissue assays.
- Non-selectivity means MT-II engages the receptor with high affinity but no subtype discrimination, so receptor-level experiments must isolate MC1R by design rather than by the ligand.
Because MT-II is non-selective, receptor-level experiments typically use MC1R-transfected cell lines, selective melanocortin antagonists, or receptor-knockout comparisons to attribute a pigmentation signal specifically to MC1R rather than to the broader melanocortin family. Structural and biophysical work on the melanocortin receptors—including recent cryo-EM structures of MC1R and MC4R bound to peptide agonists—has clarified how the peptide message sequence docks and triggers the conformational change that couples the receptor to its G protein.
What signaling pathway does Melanotan II activate through MC1R?
The defining event in MC1R pigmentation research is activation of the Gs–cAMP–PKA–CREB–MITF axis. When MT-II binds and stabilizes the active MC1R conformation, the receptor exchanges GDP for GTP on the stimulatory G protein (Gs), whose α-subunit then activates adenylyl cyclase. This raises intracellular cAMP, which activates protein kinase A. PKA phosphorylates the transcription factor CREB (cAMP response element-binding protein) at Ser-133, and phospho-CREB drives transcription of MITF (microphthalmia-associated transcription factor), the master regulator of the melanocyte differentiation program.[3]
MITF is the node that converts receptor signaling into pigment output. It transactivates the enzymatic machinery of melanogenesis:
- Tyrosinase (TYR) — the rate-limiting enzyme catalyzing tyrosine → DOPA → dopaquinone.
- Tyrosinase-related protein 1 (TYRP1) and DCT/TYRP2 — enzymes that steer the pathway toward eumelanin.
- Structural and melanosome-maturation proteins that build and traffic the pigment organelle.
Functionally, sustained MC1R signaling favors a shift from red/yellow pheomelanin toward brown/black eumelanin, the photoprotective pigment. This eumelanin bias is the mechanistic core of what afamelanotide exploits clinically and what MT-II reproduces in research models.[2]
The biochemistry of the eumelanin switch
The pheomelanin-to-eumelanin switch is where the receptor signal becomes visible chemistry. Both pigments begin from the same substrate: tyrosinase oxidizes tyrosine to dopaquinone. From there the pathway branches. In the presence of cysteine or glutathione, dopaquinone is diverted into pheomelanin (the sulfur-containing, red-yellow, poorly photoprotective—and potentially pro-oxidant—pigment). When MC1R signaling raises tyrosinase, TYRP1, and DCT activity, the flux is instead pushed toward eumelanin, the brown-black polymer that absorbs UV and scavenges free radicals. This is why MC1R is not simply a “more pigment” switch but a quality-of-pigment switch: the same total melanocyte activity can be photoprotective or photosensitizing depending on which branch dominates.[2] Individuals with loss-of-function MC1R variants are locked toward the pheomelanin branch, which is one mechanistic thread connecting red-hair genotypes to elevated melanoma risk independent of tanning behavior.
Beyond cAMP: ERK/MAPK and secondary cascades
MC1R signaling is not exclusively cAMP-driven. In melanocyte models, MC1R activation can engage the ERK/MAPK cascade, in part through cAMP-dependent mechanisms and cross-talk with the receptor tyrosine kinase c-KIT. ERK activity feeds back onto MITF, modulating its stability and activity in a context-dependent way, which is one reason MITF regulation is not a simple linear function of cAMP. The extent and biological direction of ERK, PI3K/AKT, and β-arrestin-dependent signaling downstream of MC1R remain areas of active characterization rather than settled fact, and studies of naturally occurring MC1R mutants show that cAMP, arrestin recruitment, and ERK activity can be uncoupled—evidence of biased signaling at this receptor.[6]
The MC1R signaling story also extends beyond pigment. cAMP/PKA output has been linked in melanocyte models to induction of DNA-repair capacity and antioxidant responses, which is why MC1R is sometimes framed as a genome-protective receptor. Work comparing wild-type and variant receptors has shown that intact MC1R signaling supports nucleotide-excision repair of UV-induced DNA damage and enhances cellular resistance to oxidative stress, effects that are attenuated in loss-of-function variants. In this framing, the pigment (eumelanin) and the intracellular protective program (DNA repair, antioxidant enzymes) are two arms of the same photoprotective response. These are important context but are downstream of, and mechanistically separable from, the pigmentation readout that is the focus of MT-II research—and they are studied with afamelanotide and endogenous agonists at least as much as with MT-II.

Constitutive activity and why the baseline matters
MC1R is notable among GPCRs for having measurable constitutive (ligand-independent) activity—it generates some cAMP even without agonist. Certain naturally occurring mutants alter this basal tone, and some variants change the ratio of downstream outputs rather than the total. This is why studies of MC1R mutants describe cAMP, arrestin recruitment, and ERK activity moving independently: the receptor does not have a single “activity dial.”[6] For MT-II research, the practical implication is that a superpotent agonist is being applied on top of a receptor that already has a genotype-specific baseline—so the interesting quantity is often the fold-change over baseline, not the absolute signal, and comparisons are only meaningful when the receptor allele and its constitutive tone are held constant.
What does the pigmentation evidence actually show?
The honest summary is that the mechanism is well established and internally consistent, while the compound-specific clinical evidence for Melanotan II is thin and dominated by adverse-event case reports rather than controlled trials. It is essential to separate three tiers of evidence.
Mechanistic and cell-based evidence
At the receptor and cellular level, the MC1R–cAMP–MITF–tyrosinase pathway is one of the best-mapped signaling cascades in cutaneous biology. In melanocyte cultures and melanoblast-derived systems, melanocortin stimulation reproducibly increases intracellular cAMP, upregulates tyrosinase and MITF-target gene expression, and elevates measurable melanin content, with reported increases in melanin-associated activity on the order of several-fold under controlled conditions.[3] Ex vivo human skin and pigment-cell models similarly show enhanced eumelanin synthesis and a pheomelanin-to-eumelanin shift consistent with POMC/α-MSH-mediated signaling.[2] As a superpotent, stable α-MSH mimetic, MT-II is a natural tool ligand for these assays.
Human observational and case-report data
What exists for MT-II in humans is largely a body of dermatology case reports arising from unregulated recreational tanning use, not prospective research. These consistently document generalized tanning and, importantly, changes in melanocytic nevi: darkening of existing moles, rapid eruption of new and dysplastic nevi, and changes in size and shape of pre-existing lesions.[7] Several reports describe melanoma or melanoma in situ arising in temporal association with Melanotan use.[8] Temporal association is not proof of causation, and confounding by concurrent UV/sunbed exposure is common, but the signal is coherent with the known biology and is why the pigmentation-research framing must be paired with explicit melanoma vigilance.
The afamelanotide reference standard
The instructive contrast is afamelanotide (Scenesse), the linear MC1R agonist that did undergo rigorous development. It received European Medicines Agency approval in 2014 and U.S. FDA approval in October 2019 as a controlled-release subcutaneous implant to increase pain-free light exposure in adults with erythropoietic protoporphyria (EPP), via MC1R-driven eumelanin induction.[9] In EPP, a deficiency of ferrochelatase causes phototoxic protoporphyrin IX to accumulate in the skin, so increasing epidermal eumelanin—an efficient absorber of the relevant wavelengths—raises the threshold for light-induced pain. The mechanism is the same MC1R–cAMP–eumelanin axis that MT-II activates; the difference is that afamelanotide was tested in placebo-controlled trials, is delivered as a dose-controlled implant by a clinician, and is used for a defined photoprotective indication. The existence of an approved, trial-backed MC1R agonist underscores that the pathway is therapeutically real—the next section unpacks how sharply it diverges from MT-II in selectivity, evidence, and regulatory status.
The PT-141 branch: what MC4R selectivity looks like
The Melanotan II research lineage also produced a compound that deliberately exploits the “off-target” effect. During early MT-II studies, spontaneous erections were observed as a consequence of central MC4R activation. That observation was developed into bremelanotide (PT-141), a metabolite-derived melanocortin analog optimized toward the sexual-function pathway rather than pigmentation. PT-141 is the clearest demonstration that the melanocortin receptor family is functionally modular: shifting a peptide’s receptor-subtype bias moves the biology from skin to central nervous system. For researchers characterizing the two overlapping activity profiles side by side, the combined Melanotan II and PT-141 research protocol documents the handling parameters for each.
Why do MC1R variants matter for interpreting Melanotan II research?
MC1R is one of the most polymorphic genes in the human genome, and its variants are a central reason receptor-level pigmentation research is nuanced. The red-hair-color (RHC) variants—notably R151C, R160W, and D294H—are classic loss-of-function or hypomorphic alleles that impair the receptor’s ability to raise cAMP in response to agonist.[4]
The mechanisms of loss of function differ by variant, which matters for how a ligand like MT-II behaves:
- Reduced cell-surface expression: variants such as R151C and R160W show impaired trafficking to the plasma membrane, so fewer receptors are available to bind agonist and couple to cAMP.[4]
- Impaired G-protein coupling: variants such as D294H may reach the surface but signal poorly, indicating a coupling defect rather than a trafficking defect.
- Dominant-negative behavior: several RHC variants can suppress wild-type receptor signaling when co-expressed, so heterozygous genotypes are not simply intermediate.[10]
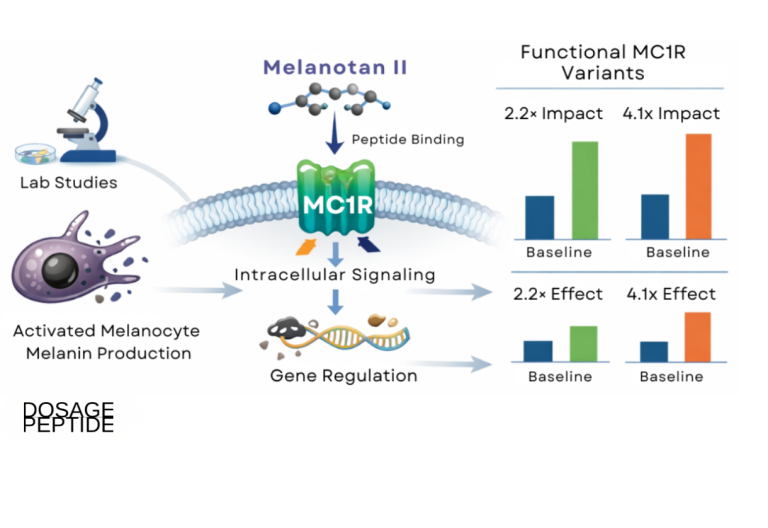
A broad review of chronic MC1R activation notes that functional MC1R variants substantially change the biological consequences of pathway stimulation, with reported differences in effect magnitude across genotypes—reinforcing that any MT-II pigmentation readout is genotype-dependent and cannot be generalized across individuals or cell lines without characterizing the receptor allele.[1] For research design, this means MC1R genotype is a variable to be controlled, not assumed.
How does Melanotan II compare with afamelanotide (Scenesse)?
Because Melanotan II and afamelanotide sit on the same α-MSH-derived branch and hit the same primary receptor, they are constantly conflated—including in casual marketing that borrows afamelanotide’s legitimacy to imply MT-II is “basically the same thing.” They are not. The differences in selectivity, evidence base, and regulatory status are the whole point.
Selectivity: targeted versus pan-melanocortin
Afamelanotide (Melanotan I) is a linear 13-residue analog that behaves as a potent and comparatively MC1R-selective agonist, with reported nanomolar affinity and cAMP EC50 values at MC1R that make it an efficient eumelanin inducer.[15] Melanotan II, by contrast, is a shorter cyclic heptapeptide that—precisely because the lactam bridge locks the core message sequence into a rigid, high-affinity conformation—binds MC1R, MC3R, MC4R, and MC5R with far less discrimination. The design trade-off is stark: MT-II’s constraint buys potency and stability but sacrifices selectivity, so it drives central MC3R/MC4R and exocrine MC5R signaling alongside pigmentation. Afamelanotide’s linear structure keeps its activity nearer the intended MC1R lane, which is one reason its clinical adverse-event profile is dominated by expected pigmentary effects rather than nausea, appetite change, and priapism.
Evidence base: placebo-controlled trials versus case reports
Afamelanotide was developed through a conventional pharmaceutical program, including randomized placebo-controlled trials that measured pain-free light exposure as a defined endpoint in EPP patients; the approved product is a dose-controlled subcutaneous implant administered by a clinician. Melanotan II has no comparable trial program. Its human record is a scattered collection of dermatology and emergency-medicine case reports arising from unregulated self-injection—valuable as safety signals, but the opposite of a controlled efficacy dataset. The gulf is not subtle: one compound has a labeled indication and a package insert; the other has a literature of adverse events.
The MC1R-selective concept has since advanced further. Dersimelagon (MT-7117), an orally administered small-molecule selective MC1R agonist, was evaluated in a randomized, placebo-controlled Phase 2 trial in EPP/XLP that significantly increased the duration of symptom-free sunlight exposure—a difference from placebo of roughly 54–63 minutes at week 16 depending on dose—via the same eumelanin-mediated mechanism, evidence that the therapeutic value lives in selective MC1R engagement, not in the pan-melanocortin promiscuity that characterizes MT-II.[16]
Regulatory status: approved medicine versus research chemical
| Attribute | Afamelanotide (Scenesse) | Melanotan II |
|---|---|---|
| Structure | Linear 13-residue α-MSH analog | Cyclic (lactam-bridged) heptapeptide |
| Receptor selectivity | Comparatively MC1R-selective | Non-selective (MC1R/MC3R/MC4R/MC5R) |
| Human evidence | Randomized placebo-controlled trials | Case reports only; no controlled trials |
| Delivery | Clinician-administered controlled-release implant | Self-injected unregulated powder |
| Regulatory status | EMA (2014) and FDA (2019) approved for EPP | Unapproved; research chemical only |
The takeaway is that afamelanotide is the correct reference standard for what a rigorously studied melanocortin drug looks like, and MT-II is the cautionary counterpoint: same pathway, same headline effect, radically different level of evidence and control. Sharing a receptor target does not make an unapproved research chemical equivalent to an approved medicine.
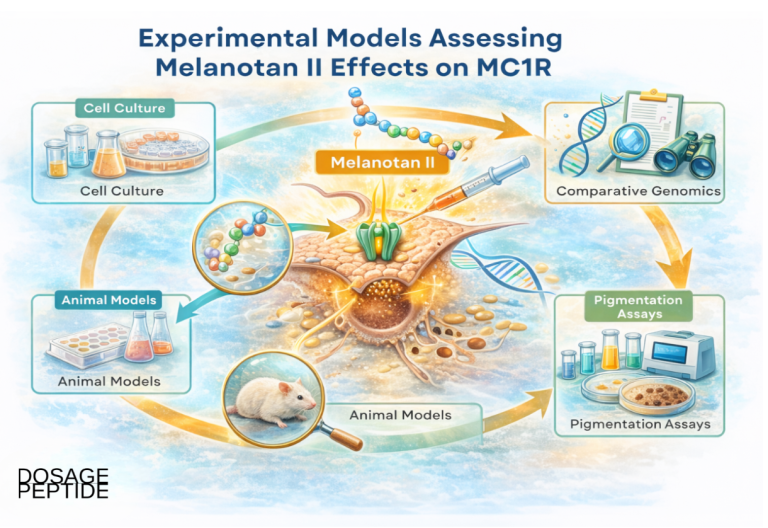
Which research models and methods are used to study MC1R signaling?
Because Melanotan II is non-selective and MC1R is polymorphic, methodology is where credible pigmentation research is won or lost. Common systems and readouts include:
| Model system | Typical readouts | Strengths / caveats |
|---|---|---|
| MC1R-transfected cell lines (e.g., HEK293) | cAMP accumulation, receptor binding, surface expression | Isolates MC1R from other MC receptors; not a full melanocyte context |
| Primary or immortalized melanocytes (human/murine) | Tyrosinase activity, MITF/TYR/TYRP1 expression, melanin content | Physiologically relevant; genotype and donor variability must be controlled |
| Melanoblast-derived lines | Differentiation, migration, melanogenic gene induction | Captures developmental biology; interpretation model-specific |
| Ex vivo human skin explants | Eumelanin synthesis, pheomelanin→eumelanin shift over 48–72 h | Preserves tissue architecture; limited assay window |
| Rodent pigmentation models | Coat/skin pigmentation, dose-response | Whole-organism context; species differences in melanocortin biology |
Rigorous MC1R attribution generally requires at least one of: a selective melanocortin antagonist, MC1R knockdown/knockout comparison, or receptor-transfected controls, so that a pigmentation signal is not silently driven by MC3R/MC4R/MC5R. Quantitative endpoints—standardized cAMP measurements, qPCR of MITF-target genes, and spectrophotometric melanin quantification—are what make results reproducible across batches and labs.
The workhorse pigmentation models in detail
A handful of model systems recur across the melanogenesis literature, each answering a different question and each with a characteristic blind spot. Understanding what each can and cannot show is essential to reading MT-II research critically.
Melanocyte culture. Primary or immortalized melanocytes (human or murine) are the most physiologically faithful in-vitro system, because they carry the full endogenous melanogenic apparatus—MC1R at native expression levels, MITF, and the tyrosinase/TYRP1/DCT enzyme set—so a melanocortin stimulus can be read out as genuine tyrosinase activity, MITF-target gene induction, and spectrophotometric melanin content. Their weakness is variability: pigmentation responses depend on donor MC1R genotype and passage number, so donor-to-donor differences must be controlled rather than assumed away.
Melanoma cell lines. The murine B16 series (notably B16-F1 and B16-F10) is the classic pigmentation screening line. B16 cells retain melanogenic capacity in culture and respond to α-MSH and other cAMP-elevating activators with a robust, reproducible rise in tyrosinase activity and melanin content via the cAMP–PKA–CREB–MITF axis—while, notably, dopachrome tautomerase can move in the opposite direction, so tyrosinase and melanin are the appropriate readouts rather than the full enzyme set.[17] This makes them the default platform for ranking pigmentation stimulators and inhibitors. The obvious caveat is that they are transformed cells: exactly because they are melanoma-derived, they are excellent for pathway kinetics but a poor model of the safety question of whether melanocortin stimulation promotes malignant transformation.
Zebrafish. Zebrafish larvae provide a rapid whole-organism, phenotype-based readout: their melanophores visibly disperse or aggregate melanosomes in response to melanocortin signaling, allowing transparent-embryo imaging and quantification without dissection. The system has been used as a bioassay for α-MSH-analog potency, and in that setting MT-II produced greater melanophore dispersion than native human α-MSH—a direct demonstration of its superpotency in a live animal, and a validation of the assay for ranking analog activity.[18] The trade-off is evolutionary distance: fish pigment biology (melanosome translocation within melanophores) is not a perfect proxy for the melanogenic, per-cell melanin-synthesis biology of mammalian skin.
Guinea pig UV models. For whole-organism induced pigmentation, the brownish/pigmented guinea pig is a long-standing in-vivo tanning model because its UV response resembles human tanning. Daily UVB exposure over several days induces clearly visible epidermal pigmentation that peaks about a week after irradiation, with increased melanocyte size, dendricity, and keratinocyte pigment—closely paralleling the human tanning timeline.[19] This makes it a useful platform for studying UV-driven, POMC/α-MSH-mediated melanogenesis in intact skin—the very loop MT-II bypasses—though species differences in melanocortin biology and interfollicular melanocyte distribution limit direct extrapolation to humans.
No single model settles a pigmentation claim. The convergent-evidence logic that credible reviews apply is to require agreement across tiers—receptor-level cAMP in a transfected line, enzyme/gene induction in melanocytes, phenotype in zebrafish or guinea pig—before treating an MT-II effect as established rather than assay-specific.
Reading a dose-response curve at MC1R
A well-designed MT-II pigmentation experiment reports more than “pigment went up.” The informative outputs are pharmacological parameters: EC50 (the concentration producing half-maximal cAMP or melanin response), Emax (the ceiling of the response), and, where antagonists are used, the rightward shift in the curve that quantifies competitive blockade. Because MT-II is superpotent, its EC50 at MC1R sits well below that of native α-MSH, which is precisely why it is a good tool for probing receptor behavior at low occupancy. Comparing curves across MC1R genotypes—wild-type versus R151C, R160W, or D294H—converts the abstract idea of “variant loss of function” into a measured reduction in Emax or a rightward EC50 shift, and it is the standard way variant severity is ranked.
Controls that separate signal from artifact
Several controls recur in credible pigmentation studies. A vehicle/diluent control distinguishes peptide effect from the reconstitution buffer. A time-matched control accounts for spontaneous melanization in culture. A receptor-negative line (or MC1R knockout) confirms the readout collapses when the target is removed. An antagonist arm (for example, agouti-signaling protein, the endogenous inverse agonist at MC1R) confirms the signal is receptor-mediated and reversible. Where these controls are absent, an apparent MT-II effect cannot be confidently attributed to MC1R, and this is a frequent weakness in low-quality secondary literature that should be read skeptically.
Handling, reconstitution, and research-dosing context
Melanotan II is supplied as a lyophilized (freeze-dried) powder for laboratory use and is not approved for human use in any jurisdiction; it is sold as a research chemical only. Nothing in this section is dosing guidance for human administration—it describes how the compound is handled in experimental settings so that pigmentation research is reproducible.
In research workflows, lyophilized MT-II is typically reconstituted with bacteriostatic water to a defined stock concentration, kept cold, and protected from repeated freeze-thaw cycles that degrade peptides. Accurate reconstitution is the single largest source of avoidable concentration error in peptide assays: the difference between intending a 1 mg/mL stock and accidentally preparing 0.5 mg/mL is a two-fold error propagated through every downstream dose-response point. The Dosage Peptide peptide reconstitution guide and the reconstitution dosage calculator walk through the diluent-volume math that fixes your working concentration, and the compound-specific Melanotan II 10 mg vial dosage protocol collects the handling parameters used in research characterization.
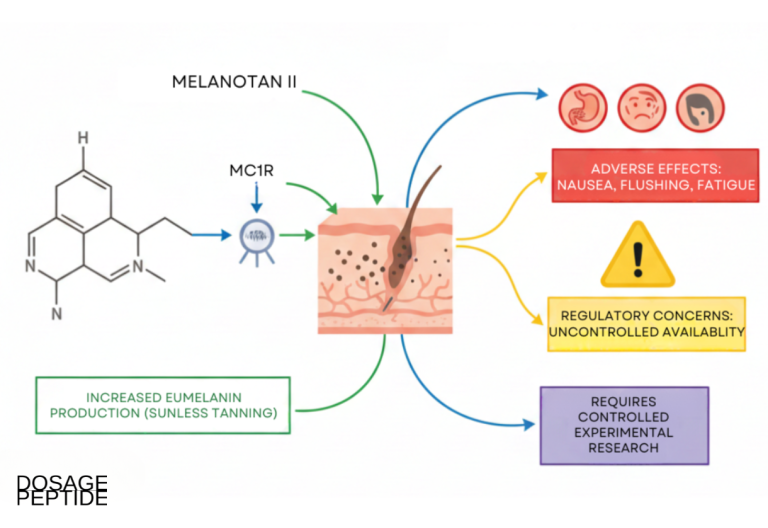
What are the safety signals and open questions?
Documented safety signals
The adverse-event profile associated with Melanotan is well described in the dermatology literature and follows directly from its pharmacology:
- Melanocytic and melanoma concern: darkening of existing nevi, eruptive and dysplastic new nevi, and case reports of melanoma arising in temporal association with use—the most serious signal and the reason MC1R pigmentation research must be paired with lesion surveillance.[11]
- Off-target melanocortin effects: because MT-II is non-selective, MC3R/MC4R engagement produces nausea, flushing, appetite suppression, and spontaneous penile erections—the very effects that spun off PT-141/bremelanotide as a separate line.
- Cardiovascular and systemic signals: case reports and pharmacovigilance summaries describe blood-pressure changes, and the broad melanocortin activity means systemic effects cannot be assumed to be pigment-limited.
- Product-quality risk: as an unregulated research chemical, MT-II sold outside controlled supply chains carries risks of impurity, mislabeling, bacterial contamination, and non-sterile handling—variables that also confound any research interpretation.[7]
The melanocytic signal deserves special emphasis because it is mechanistically expected, not incidental. A ligand that drives melanocyte proliferation, migration, and pigment activity through MC1R is, by construction, stimulating the same cell population from which melanoma arises. That does not establish that MT-II is carcinogenic—the case reports are confounded and uncontrolled—but it means the null hypothesis should be treated cautiously, and it is why the dermatology literature consistently pairs any discussion of Melanotan with the ABCDE mole-monitoring warning signs.
What the case-report literature actually documents
Because MT-II has no controlled human trials, its documented human harms come from published case reports—which, while individually anecdotal, cluster into coherent, pharmacology-consistent patterns. It is worth being specific about what has actually been reported, because the specifics are more sobering than a generic “side effects” note.
Changing and eruptive melanocytic nevi with melanoma concern. The dermatology literature contains repeated, well-documented reports of nevus change after Melanotan use. Cardones and Grichnik described α-MSH-analog-induced eruptive nevi and the transformation of existing nevi with atypical clinical and histologic features in a patient predisposed to melanoma, concluding that these peptides can drive proliferation of neoplastic melanocytic cells in susceptible individuals.[20] A separate report documented a thin (0.3 mm) melanoma arising in a pre-existing abdominal nevus three months after Melanotan injections.[8] Investigators are careful that causation is not established—affected patients typically also had fair phototype, many nevi, personal/family melanoma history, and heavy UV/sunbed exposure—but the biological plausibility, combined with the reproducibility of the nevus-change observation, is exactly why lesion surveillance is non-negotiable in this context.
Rhabdomyolysis and systemic sympathomimetic toxicity. A published case describes a 39-year-old man who injected 6 mg of internet-sourced Melanotan II—several times a typical starting amount—and presented with diffuse myalgia, sweating, anxiety, tachycardia, and hypertension. His creatine phosphokinase rose from roughly 1,760 IU/L on admission to over 17,000 IU/L within 12 hours, confirming rhabdomyolysis, with mildly elevated troponin and renal dysfunction; he required ICU-level intravenous fluid and bicarbonate therapy before recovering.[21] The episode illustrates two compounding hazards: broad melanocortin/sympathomimetic activity, and the dosing imprecision inherent to self-injected, unregulated powder.
Priapism. Because MT-II activates central MC4R—the same mechanism deliberately harnessed by bremelanotide—prolonged, painful erection is a recognized emergency presentation. A published case reports a long-term Melanotan II user who developed a sustained ischemic priapism lasting about 30 hours after a subcutaneous injection, requiring escalating urological intervention: cavernosal aspiration and intracavernosal phenylephrine failed to achieve detumescence, and he ultimately underwent operative penoscrotal decompression.[22] Priapism beyond four hours is a urological emergency that can cause permanent erectile damage—a direct, foreseeable consequence of the very MC4R pharmacology that makes MT-II interesting to melanocortin researchers.
These reports share a through-line: each harm is a predictable extension of MT-II’s known receptor pharmacology (non-selective melanocortin agonism plus sympathomimetic activation), amplified by unregulated sourcing and self-dosing. That is the opposite of a reassuring safety picture, and it is the empirical backbone of the “not approved for human use” framing that runs through this article.
Genuine research gaps
Several questions remain genuinely unresolved and shape research priorities:
- Variant-specific responsiveness: systematic profiling of MT-II signaling across the full spectrum of MC1R polymorphisms—beyond the classic RHC alleles—is incomplete.
- In vivo receptor selectivity: discriminating MC1R- from MC4R-driven effects in whole organisms, and mapping tissue-specific signaling, remains under-characterized beyond rodent systems.
- Biased and long-term signaling: the balance between cAMP, ERK, and β-arrestin outputs—and the consequences of sustained activation in advanced models such as iPSC-derived melanocytes and 3D skin organoids—is largely unexplored.
- Causality of the melanoma signal: whether MT-II independently promotes malignant transformation, or primarily amplifies UV-driven risk in predisposed individuals, has not been resolved by controlled study.
The overarching honest conclusion is that Melanotan II is a valuable mechanistic tool for dissecting MC1R signaling, but it is not a validated therapeutic and should be understood through the lens of its approved, trial-backed cousin afamelanotide when weighing what melanocortin pharmacology can and cannot claim.
Frequently Asked Questions
How does Melanotan II activate MC1R at the molecular level?
Melanotan II binds the orthosteric pocket of MC1R and stabilizes the receptor’s active conformation. This triggers Gs-mediated activation of adenylyl cyclase, raising cAMP, which activates PKA. PKA phosphorylates CREB, driving MITF transcription, and MITF upregulates tyrosinase and related enzymes that increase eumelanin synthesis. The cyclic lactam structure of Melanotan II makes this activation potent and sustained in research models.
Is Melanotan II the same as afamelanotide (Scenesse)?
No. Both are α-MSH-derived melanocortin agonists, but afamelanotide (Melanotan I / Scenesse) is a linear peptide that completed clinical development and is approved for erythropoietic protoporphyria. Melanotan II is a cyclic, non-selective analog that is unapproved and sold only as a research chemical. They share the MC1R target but differ in selectivity, evidence base, and regulatory status, and are not interchangeable.
Why does Melanotan II cause side effects beyond tanning?
Melanotan II is a non-selective melanocortin agonist. In addition to MC1R (pigmentation), it activates MC3R, MC4R, and MC5R. MC4R activation in the central nervous system drives appetite suppression and penile erection, while broad melanocortin signaling contributes to nausea and flushing. These off-target effects are inherent to its non-selective pharmacology, not incidental impurities.
What is the connection between Melanotan II and melanoma risk?
Dermatology case reports link Melanotan use to darkening of existing moles, eruption of new and dysplastic nevi, and melanoma arising in temporal association with use. Because Melanotan II stimulates melanocyte proliferation and pigment activity, and because users often combine it with UV exposure, the biology is coherent with the signal. Causation is not proven, but the concern is significant enough to warrant lesion surveillance.
Why do MC1R variants change how Melanotan II signals?
Red-hair-color MC1R variants such as R151C, R160W, and D294H are loss-of-function or hypomorphic. Some reduce receptor trafficking to the cell surface; others impair G-protein coupling; several act as dominant negatives. All reduce cAMP output for a given agonist stimulus, so the pigmentation response to Melanotan II is genotype-dependent and cannot be generalized without characterizing the receptor allele in the model.
What research models are used to study MC1R signaling?
Common systems include MC1R-transfected cell lines for isolated receptor signaling, primary and immortalized melanocytes for melanogenic readouts, melanoblast-derived lines, ex vivo human skin explants, and rodent pigmentation models. Because Melanotan II is non-selective, credible studies use selective antagonists, receptor knockdowns, or transfected controls to attribute a signal specifically to MC1R rather than to other melanocortin receptors.
Is Melanotan II approved or legal for human use?
No. Melanotan II is not approved by the FDA, EMA, or other major regulators for any human indication and is supplied strictly as a research chemical for laboratory investigation of melanocortin signaling. It is not intended for clinical, diagnostic, or therapeutic use in humans or animals. The approved MC1R agonist in this class is afamelanotide (Scenesse), for erythropoietic protoporphyria only.
What is the difference between eumelanin and pheomelanin in MC1R research?
Both pigments start from tyrosinase-generated dopaquinone. Eumelanin is the brown-black, UV-absorbing, photoprotective polymer; pheomelanin is the sulfur-containing red-yellow pigment that is poorly protective and can be pro-oxidant. Strong MC1R signaling pushes flux toward eumelanin. This quality shift, not just total pigment quantity, is why MC1R function correlates with photoprotection and why loss-of-function variants raise melanoma risk.
How is Melanotan II handled and reconstituted in a research setting?
Melanotan II ships as a lyophilized powder and is reconstituted with bacteriostatic water to a defined stock concentration for laboratory use, then kept cold and protected from freeze-thaw cycles. Accurate diluent-volume math is critical because concentration errors propagate through every dose-response point. This is handling context for research reproducibility only, not human dosing guidance.
Where does α-MSH come from in the body?
α-MSH is one of several peptides cut from a single precursor protein, proopiomelanocortin (POMC), by tissue-specific prohormone convertases. In the skin, keratinocytes and melanocytes both express POMC and its processing enzymes, and ultraviolet B exposure upregulates POMC and the release of α-MSH, which then acts on neighboring melanocytes’ MC1R to raise eumelanin. Melanotan II is a synthetic, stabilized surrogate for this endogenous α-MSH, which is why it can drive pigmentation without a UV trigger.
Has Melanotan II caused documented emergencies in case reports?
Yes. Published case reports include rhabdomyolysis with sympathomimetic toxicity and renal dysfunction after a high self-injected dose, and prolonged ischemic priapism requiring urological intervention—both consistent with Melanotan II’s non-selective melanocortin and MC4R pharmacology. There are also repeated reports of changing or eruptive melanocytic nevi and melanoma arising in temporal association with use. These are individual case reports, not controlled data, but they are coherent with the known biology and underscore that Melanotan II is an unapproved research chemical, not a therapeutic.
Is dersimelagon the same kind of drug as Melanotan II?
No. Dersimelagon (MT-7117) is an orally administered, small-molecule selective MC1R agonist evaluated in placebo-controlled trials for erythropoietic and X-linked protoporphyria. Like afamelanotide, it demonstrates that the therapeutic value of this pathway lies in selective MC1R engagement, not in the non-selective, multi-receptor activity of Melanotan II. Dersimelagon and afamelanotide are investigational or approved medicines developed through formal trials; Melanotan II is an unapproved research chemical with no comparable program.
Which animal models best mirror human tanning for pigmentation research?
For UV-induced tanning in intact skin, the pigmented guinea pig is a long-used in-vivo model because its UVB response—increased melanocyte size, dendricity, and epidermal pigment peaking about a week after exposure—parallels human tanning. Zebrafish larvae offer a faster whole-animal readout via melanophore melanosome dispersion and have been used to rank α-MSH-analog potency, with Melanotan II outperforming native α-MSH. Neither is a perfect human proxy, so credible conclusions require convergence across cell-based and whole-organism models.
References
- Böhm, M., et al. (2025). An overview of the benefits and risks of chronic melanocortin-1 receptor activation. Journal of the European Academy of Dermatology and Venereology, 39(1), 39–51.
- Maranduca, M. A., et al. (2019). Synthesis and physiological implications of melanic pigments (review). Oncology Letters / PMC6444329.
- Niu, C., & Aisa, H. A. (2017). Upregulation of melanogenesis and tyrosinase activity: potential agents for vitiligo. Molecules, 22(8), 1303.
- Beaumont, K. A., et al. (2005). Altered cell surface expression of human MC1R variant receptor alleles associated with red hair and skin cancer risk. Human Molecular Genetics, 14(15), 2145–2154.
- Hadley, M. E., & Dorr, R. T. (2006). Melanocortin peptide therapeutics: historical milestones, clinical studies and commercialization. Peptides, 27(4), 921–930 (PMID: 16426078).
- Benned-Jensen, T., Mokrosinski, J., & Rosenkilde, M. M. (2011). The E92K melanocortin 1 receptor mutant induces cAMP production and arrestin recruitment but not ERK activity, indicating biased constitutive signaling. PLoS ONE, 6(9), e24644.
- DermNet NZ. Melanotan II — overview, effects and safety concerns.
- Paurobally, D., Jason, F., Dezfoulian, B., & Nikkels, A. F. (2011). Melanotan-associated melanoma. British Journal of Dermatology, 164(6), 1403–1405.
- Drugs.com. Scenesse (afamelanotide) FDA approval history.
- Beaumont, K. A., et al. (2007). Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Human Molecular Genetics (PMID: 17616515).
- Eruptive dysplastic nevi following Melanotan use. Actas Dermo-Sifiliográficas.
- Rousseau, K., et al. (2007). Proopiomelanocortin (POMC), the ACTH/melanocortin precursor, is secreted by human epidermal keratinocytes and melanocytes and stimulates melanogenesis. FASEB Journal / PMC2253185.
- Schauer, E., et al. (1994/1996). Production and release of proopiomelanocortin (POMC)-derived peptides by human melanocytes and keratinocytes in culture: regulation by ultraviolet B. Journal of Clinical Investigation (PMID: 8781560).
- Chen, W., et al. (1997). Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell, 91(6), 789–798 (PMID: 9413988).
- Wensink, D., Wagenmakers, M. A. E. M., & Langendonk, J. G. (2021). Afamelanotide for prevention of phototoxicity in erythropoietic protoporphyria. Expert Review of Clinical Pharmacology, 14(2), 151–160.
- Balwani, M., et al. (2023). Dersimelagon in erythropoietic protoporphyrias. New England Journal of Medicine, 388(15), 1376–1385 (PMID: 37379150).
- Martínez-Liarte, J. H., Solano, F., García-Borrón, J. C., Jara, J. R., & Lozano, J. A. (1992). α-MSH and other melanogenic activators mediate opposite effects on tyrosinase and dopachrome tautomerase in B16/F10 mouse melanoma cells. Journal of Investigative Dermatology, 99(4), 435–439 (PMID: 1328399).
- Zebrafish bioassay for screening therapeutic candidates based on melanotrophic activity. (2021). International Journal of Molecular Sciences, 22(17), 9313.
- Differential analysis of experimental hypermelanosis induced by UVB, PUVA, and allergic contact dermatitis using a brownish guinea pig model. (1986). Archives of Dermatological Research (PMID: 3753033).
- Cardones, A. R., & Grichnik, J. M. (2009). α-Melanocyte-stimulating hormone-induced eruptive nevi. Archives of Dermatology, 145(4), 441–444 (PMID: 19380666).
- Nelson, M. E., & Bryant, S. M. (2012). Melanotan II injection resulting in systemic toxicity and rhabdomyolysis. Clinical Toxicology, 50(10), 1169–1173.
- Melanotan tanning injection: a rare cause of priapism. (2021). Sexual Medicine, 9(1), 100298.