The question in this article’s title reads like a settled clinical fact: that glucagon-like peptide-1 (GLP-1) receptor agonists do regulate lipid metabolism in atherogenic dyslipidemia, and that we simply need to describe the machinery. The honest answer is more layered. The GLP-1 receptor-agonist class is very real and, in several members, very well studied — semaglutide and tirzepatide (a dual GIP/GLP-1 agonist) are approved medicines with large outcome trials behind them. But the specific claim embedded in the title — that GLP-1 pathways regulate the particular constellation of lipid abnormalities called atherogenic dyslipidemia in a mechanistically defined, treatment-grade way — is better described as an active, still-maturing research question than as an established indication.1
None of the marketed GLP-1 receptor agonists is approved to treat dyslipidemia. Their lipid effects are consistently reported as secondary or exploratory endpoints, and the mechanistic story that connects receptor activation to triglyceride-rich lipoprotein handling is assembled largely from cell culture, rodent models, small kinetic studies in humans, and post-hoc analyses of trials designed for glycemic or weight outcomes.2 That does not make the biology uninteresting — it is genuinely compelling — but it means the correct framing is: through what mechanisms might GLP-1 receptor signaling influence the lipid abnormalities that drive atherosclerosis, and how strong is the evidence for each proposed step?
This article walks through that question in the spirit of DosagePeptide.com: evidence-cautious, primary-source-grounded, and careful to separate what is approved and demonstrated from what remains a hypothesis. It is written for research and educational purposes only, and it is not medical advice. Where it describes doses, handling, or reconstitution, it does so in the narrow context of how these compounds are studied — not as guidance for human use.
What GLP-1 receptor agonists are, and the origin of the lipid question
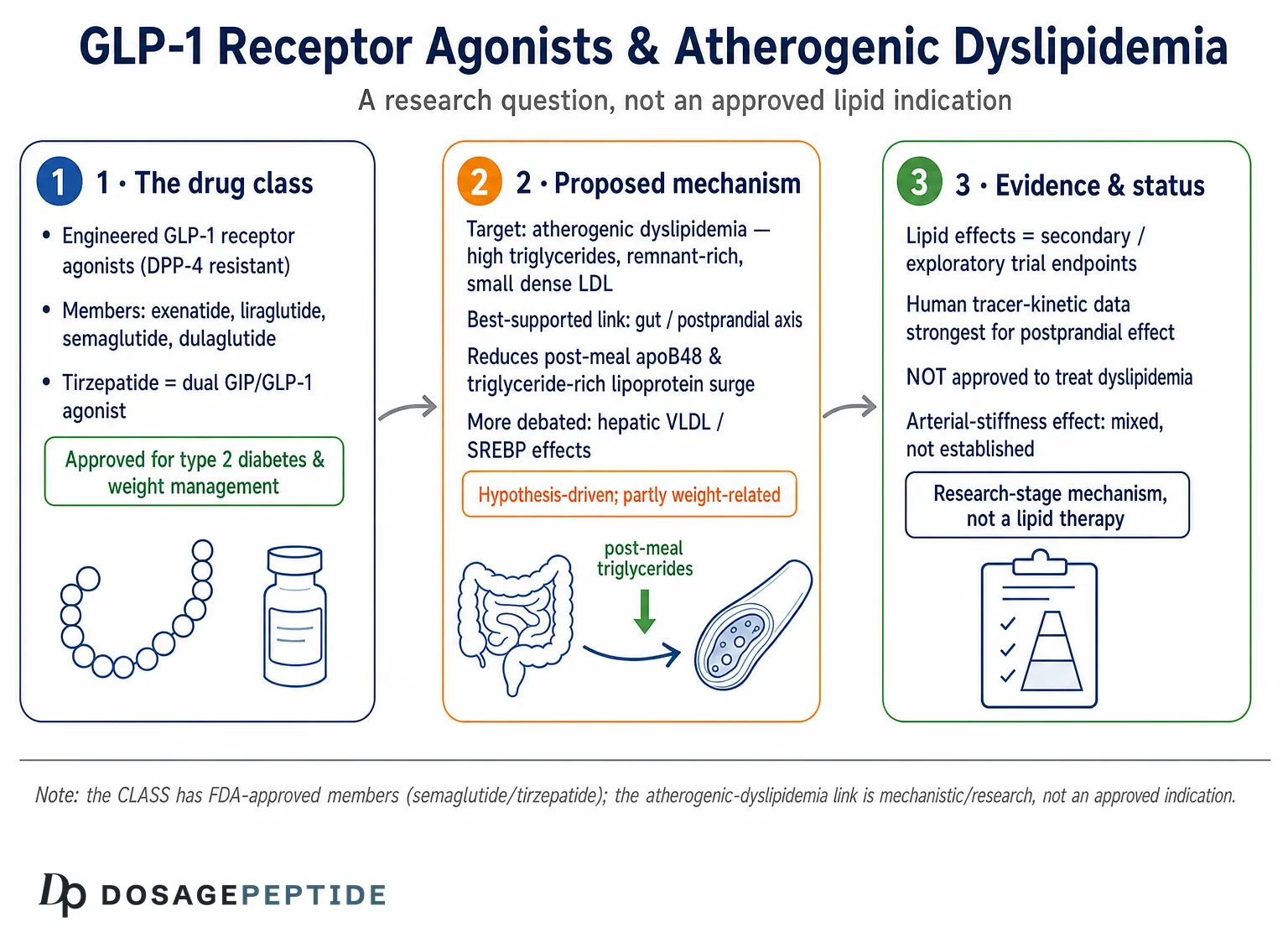
GLP-1 is an incretin hormone released from enteroendocrine L-cells of the distal small intestine and colon in response to nutrient ingestion. Native GLP-1 is a 30- or 31-amino-acid peptide cleaved from proglucagon, and its defining physiological jobs are glucose-dependent stimulation of insulin secretion, suppression of glucagon, slowing of gastric emptying, and promotion of satiety through central pathways.1 Because native GLP-1 is degraded within minutes by the enzyme dipeptidyl peptidase-4 (DPP-4), it has no practical value as a drug in unmodified form. The pharmaceutical class known as GLP-1 receptor agonists solves this by engineering DPP-4 resistance and albumin binding or fatty-acid acylation to extend half-life — exenatide and lixisenatide as shorter-acting agents, then liraglutide (daily) and the long-acting once-weekly agents semaglutide and dulaglutide.1 Tirzepatide extends the concept by co-agonizing the GIP receptor, and investigational triple agonists such as retatrutide add glucagon-receptor activity.2
The lipid question arose almost incidentally. As these agents moved through diabetes and obesity programs, investigators noticed that fasting and post-meal lipid parameters — particularly triglycerides and triglyceride-rich lipoproteins — tended to fall alongside glucose and weight.3 That observation is important because atherogenic dyslipidemia is not primarily a high-LDL-cholesterol problem; it is a triglyceride-centered, remnant-rich, small-dense-LDL phenotype for which existing therapies (statins, ezetimibe) are only partially effective. If GLP-1 pathways genuinely touched the machinery that produces and clears triglyceride-rich lipoproteins, that would be mechanistically distinct from statin action and therefore worth understanding.4
It is essential to be precise about what “GLP-1 receptor agonist” means when discussing lipids, because the class is heterogeneous. The GLP-1 receptor is a class B G-protein-coupled receptor expressed in pancreatic islets, the central nervous system, the stomach, the heart and vasculature, and — at debated levels — in the intestine and liver.5 Whether a given lipid effect is direct (receptor on a metabolically relevant cell) or indirect (secondary to weight loss, improved insulin sensitivity, reduced food intake, or slowed nutrient delivery) is one of the central unresolved issues in the field, and different studies reach different conclusions.6 Readers exploring the broader landscape of these compounds may find the site’s peptide dosage index useful for orienting which specific agents are being discussed in the research literature.
So the origin of this article’s question is empirical: a repeatable association between GLP-1 receptor agonism and improved lipid parameters, observed across multiple compounds and trials, that invites a mechanistic explanation. The rest of this piece examines the proposed mechanisms and, critically, the strength of evidence supporting each — because an association at the population level does not by itself establish that a receptor “regulates” a metabolic pathway.
Atherogenic dyslipidemia: defining the target phenotype
To evaluate whether GLP-1 pathways plausibly “regulate lipid metabolism in atherogenic dyslipidemia,” it helps to be exact about what that phenotype is. Atherogenic dyslipidemia — sometimes called diabetic dyslipidemia or the lipid triad — is characterized by elevated fasting and postprandial triglycerides, reduced high-density lipoprotein cholesterol (HDL-C), and a preponderance of small, dense low-density lipoprotein (LDL) particles, often with only modestly elevated or even normal LDL-cholesterol concentration.4 The unifying upstream driver is overproduction of apolipoprotein B (apoB)-containing lipoproteins — hepatic very-low-density lipoprotein (VLDL, carrying apoB100) and intestinal chylomicrons (carrying apoB48) — combined with impaired clearance of the resulting remnant particles.7
Why does this pattern matter more than LDL-cholesterol alone? Because the number of atherogenic particles, not just their cholesterol cargo, tracks with arterial risk. Small dense LDL penetrates the arterial intima more readily, is more susceptible to oxidation, and is cleared more slowly; triglyceride-rich remnant particles are taken up directly by intimal macrophages to form foam cells.4 This is the phenotype that persists in many people despite well-controlled LDL-cholesterol on a statin — the so-called residual risk. It is precisely because atherogenic dyslipidemia is a triglyceride- and remnant-centered problem that a therapy acting on triglyceride-rich lipoprotein production or clearance would be mechanistically interesting.3
The metabolic context is equally important. Atherogenic dyslipidemia clusters with insulin resistance, visceral adiposity, and hepatic steatosis. In insulin-resistant states, the liver fails to suppress VLDL output after meals and engages in excess de novo lipogenesis, while adipose lipolysis floods the portal circulation with free fatty acids that become VLDL substrate.8 The intestine, too, over-secretes apoB48 particles in insulin resistance. Because GLP-1 receptor agonists reduce weight, reduce hepatic fat, and improve insulin sensitivity, any lipid benefit could plausibly flow through these upstream corrections rather than through a dedicated “lipid-regulating” action of the receptor.6 Disentangling the two is one of the central methodological challenges discussed later in this article.
Framing the target this way clarifies the standard of evidence. To claim that GLP-1 pathways regulate atherogenic dyslipidemia, one would ideally show (a) measurable, reproducible changes in the defining lipid parameters, (b) a plausible molecular mechanism at a metabolically relevant tissue, (c) an effect that is at least partly independent of weight loss, and (d) linkage to clinical outcomes. As the following sections show, the field has partial support for (a), (b), and — more recently — (c), but the causal chain to (d) through lipids specifically remains inferential.9
Molecular mechanism I: hepatic lipogenesis, VLDL, and cholesterol synthesis
The liver is the most-studied candidate site for GLP-1 effects on lipids, and also the most contested, because the density of functional GLP-1 receptors on adult hepatocytes is debated. Nonetheless, a consistent body of rodent and cell work reports that pharmacological GLP-1 receptor agonism reduces hepatic VLDL-triglyceride production and blunts de novo lipogenesis. In one frequently cited study, GLP-1 receptor agonism ameliorated hepatic VLDL overproduction and de novo lipogenesis in an insulin-resistant model, an effect the authors attributed partly to reduced hepatic lipid substrate and altered expression of lipogenic enzymes.8
A proposed molecular node is the sterol regulatory element-binding protein (SREBP) family of transcription factors, master regulators of fatty-acid and cholesterol biosynthesis. SREBP-1c drives fatty-acid and triglyceride synthesis, while SREBP-2 governs cholesterol synthesis and LDL-receptor expression.10 In cultured hepatocytes exposed to palmitic acid, GLP-1 receptor agonists have been reported to inhibit SREBP-2 via a SIRT6-AMPK pathway, reducing cholesterol synthesis while enhancing hepatocyte cholesterol uptake — a mechanistic sketch that, if it holds in vivo, would connect receptor activation to cholesterol homeostasis.11 AMP-activated protein kinase (AMPK) is an attractive intermediary because it is a cellular energy sensor that phosphorylates and inhibits SREBP cleavage, dampening lipogenic and cholesterogenic transcription.10
An important caveat is that many of these hepatic effects may be indirect. Because adult hepatocyte GLP-1 receptor expression is low or difficult to detect, several investigators argue that the liver responds to GLP-1 agonism through changes in whole-body energy balance, reduced portal free-fatty-acid flux, improved insulin action, and neural (parasympathetic/vagal) signaling rather than through direct hepatocyte receptor engagement.8 A 2025 mechanistic paper even proposed that intestinal L-cell mechanoreception regulates hepatic lipid synthesis via GLP-1 release, positioning the gut — not the liver — as the primary sensor.12 The distinction matters: an indirect effect is real but is not the same as the receptor “regulating” hepatic lipid metabolism in the strict sense the title implies.
The net hepatic picture from preclinical work is therefore: GLP-1 receptor agonism reliably lowers liver fat and VLDL output in insulin-resistant rodents, plausibly engaging SREBP/AMPK biology, but the degree to which this is a direct hepatocyte program versus a downstream consequence of systemic metabolic improvement is unresolved.6 This uncertainty is not a weakness of the compounds — the fat and VLDL reductions are real — but it is a crucial limit on how confidently one can label GLP-1 a hepatic lipid regulator. For readers tracking the specific molecules in which these hepatic effects have been examined, the semaglutide and tirzepatide protocol pages, such as the semaglutide dosage protocol, identify the exact agents used in the underlying studies.
Molecular mechanism II: the intestinal axis and postprandial lipemia
If the hepatic story is contested, the intestinal story is arguably the most mechanistically robust link between GLP-1 signaling and atherogenic lipids — and it is centered on postprandial rather than fasting lipemia. After a fatty meal, enterocytes assemble dietary lipid into apoB48-containing chylomicrons; in insulin-resistant states this process is exaggerated, producing a surge of triglyceride-rich, atherogenic remnant particles. Multiple human kinetic studies show that GLP-1 receptor agonists blunt this surge.13
In a randomized study in people with type 2 diabetes, liraglutide reduced postprandial hyperlipidemia by both increasing apoB48 catabolism and reducing apoB48 production — a dual effect on synthesis and clearance measured by stable-isotope tracer kinetics, which is among the more direct human demonstrations available.13 A separate acute human study of exenatide is particularly informative on mechanism: it suppressed the production rate of intestinal apoB48-containing lipoproteins in healthy men under carefully clamped conditions, and the authors interpreted the effect as at least partly independent of both weight change and gastric emptying — pointing toward a more direct action on enterocyte lipoprotein assembly than the “slower stomach” explanation alone would predict.14 Broader reviews likewise report that GLP-1 receptor agonists consistently lower postprandial apoB48 and chylomicron triglyceride, and one frequently proposed contributor is reduced apolipoprotein C-III, an inhibitor of lipoprotein lipase whose fall would accelerate remnant clearance — though it should be stressed that the apoC-III step is a mechanistic hypothesis rather than a firmly established link, and it is cited here as a candidate pathway, not a proven one.14 A 2024 analysis of tirzepatide similarly found improved post-challenge lipid responses in type 2 diabetes.15
How does the receptor reach the intestine? Two non-exclusive routes are proposed. The first is slowed gastric emptying and reduced fat absorption, which mechanically reduces the substrate delivered to enterocytes. The second, more interesting route is neural: preclinical work indicates that GLP-1 attenuates intestinal fat absorption and chylomicron production via vagal afferent nerves originating in the portal vein, and that central GLP-1 signaling suppresses intestinal triglyceride-rich lipoprotein secretion.16 This gut-brain-gut axis would let circulating agonists influence enterocyte secretion without requiring abundant enterocyte receptors.17
There is, however, a genuinely humbling complication in the endocrinology here. GLP-1 and its sibling proglucagon product GLP-2 have opposing effects on intestinal lipoprotein output — GLP-2 stimulates chylomicron secretion while GLP-1 suppresses it — and some human physiology studies suggest GLP-2’s stimulatory action may predominate in normal and insulin-resistant states, with endogenous GLP-1 contributing only modestly to postprandial lipemia.18 This is a useful reminder that pharmacological agonism at supraphysiologic exposure is not the same as the physiological role of the endogenous hormone. The intestinal evidence for the drug class is strong; the claim that endogenous GLP-1 is a dominant native regulator of postprandial lipids is weaker and disputed.18
It is also worth noting where this intestinal effect sits in the daily physiology of a person with atherogenic dyslipidemia. Humans spend most of waking hours in a postprandial or fed state, so an agent that consistently trims the height and duration of post-meal triglyceride and remnant excursions could, in aggregate, meaningfully lower cumulative arterial exposure to atherogenic particles — even if fasting values move only modestly. This is one reason the postprandial axis is attractive mechanistically: it targets a window that fasting-focused lipid panels routinely miss, and it aligns with the observation that remnant cholesterol is an increasingly recognized, partly independent contributor to atherosclerotic risk.4 The corollary, however, is that standard fasting lipid profiles may under-represent whatever benefit exists, which complicates both trial interpretation and any claim about magnitude.14
On balance, the intestinal axis is where the phrase “GLP-1 pathways regulate lipid metabolism” comes closest to being defensible: there are human tracer-kinetic data, a plausible neural mechanism, and reproducibility across agents. Even here, though, the effect is best described as attenuation of postprandial excursions rather than wholesale reprogramming of lipid metabolism.14
What the human lipid evidence actually shows
Stepping back from mechanism to measured outcomes, what happens to the lipid panel when humans receive GLP-1 receptor agonists? The consistent finding across trials and meta-analyses is a modest but reproducible improvement, most pronounced for triglycerides. In the pivotal obesity trial STEP 1, subcutaneous semaglutide 2.4 mg was associated with roughly 12–17% reductions in triglycerides and single-digit reductions in LDL-cholesterol and total cholesterol from baseline.19 A systematic review and meta-analysis of randomized trials in overweight and obese non-diabetic adults reached similar conclusions: semaglutide reduced triglycerides, total cholesterol, and LDL-cholesterol, with small and variable changes in HDL-cholesterol.20
Tirzepatide, with added GIP-receptor activity, tends to show somewhat larger triglyceride and VLDL improvements. In the head-to-head SURPASS-2 trial in type 2 diabetes, tirzepatide 15 mg reduced triglycerides more than semaglutide 1.0 mg (on the order of 24% versus 17%) and produced greater VLDL-cholesterol reductions and HDL-cholesterol increases; a meta-analysis of tirzepatide trials confirmed reductions in triglycerides, total and LDL cholesterol with modest HDL gains.21 Oral semaglutide has likewise been shown to lower fasting and postprandial triglycerides relative to placebo.22
The following table summarizes representative directions and rough magnitudes reported in the literature. These figures are approximate, vary by agent, dose, population, and baseline, and should not be read as guaranteed or clinically prescriptive effects.
| Lipid parameter | Typical reported direction | Approximate magnitude (varies widely) | Evidence character |
|---|---|---|---|
| Fasting triglycerides | Decrease | ~10–25% | Consistent across RCTs/meta-analyses20 |
| Postprandial TG / apoB48 | Decrease | Meal-dependent, often substantial | Human tracer-kinetic studies13 |
| LDL-cholesterol | Small decrease | ~3–8% | Modest, sometimes non-significant19 |
| Total cholesterol | Small decrease | ~5–10% | Consistent but modest21 |
| HDL-cholesterol | Small increase or no change | ~0–8% | Variable20 |
| VLDL-cholesterol | Decrease | Larger with tirzepatide | Post-hoc / secondary endpoints21 |
Two honest qualifications frame these numbers. First, the effect sizes are meaningful for triglycerides but modest for LDL-cholesterol — these are not statin-magnitude LDL reductions, and no GLP-1 receptor agonist is a substitute for LDL-lowering therapy where that is indicated.19 Second, in most trials the lipid endpoints were secondary or exploratory, and a large share of the improvement tracks with weight loss. A key recent nuance, discussed next in the comparisons section, is emerging evidence that a portion of the lipid change occurs independently of weight reduction — a finding that strengthens, without proving, the case for a direct metabolic action.23
The overall verdict at the human-outcome level is that GLP-1 receptor agonists reproducibly nudge the atherogenic lipid phenotype in a favorable direction, with the clearest signal on triglyceride-rich lipoproteins. Whether that nudge is large enough to matter clinically, and whether it is causally upstream of cardiovascular benefit, are separate questions addressed below.
Cardiovascular outcomes, arterial stiffness, and the vessel wall
The strongest clinical data for this class are cardiovascular outcome data — but these do not, by themselves, prove that lipids are the mechanism. The landmark SELECT trial randomized 17,604 adults with established cardiovascular disease and overweight or obesity but without diabetes to semaglutide 2.4 mg or placebo on top of standard care, and reported a roughly 20% reduction in major adverse cardiovascular events (cardiovascular death, non-fatal myocardial infarction, or stroke).24 Crucially, participants were already receiving lipid-lowering therapy, the benefit emerged early, and mediation analyses suggest weight change accounted for only a fraction of the effect — implying multiple contributing mechanisms, of which lipid modification is at most one.24
This is the central interpretive point for the article’s question. GLP-1 receptor agonists clearly reduce cardiovascular events in appropriate populations, and their broader cardioprotective profile is recognized in cardiology guidance for people with type 2 diabetes and atherosclerotic disease.9 But the pathways proposed to deliver that benefit are numerous — anti-inflammatory effects on the vessel wall, blood-pressure reduction, improved endothelial function, weight and glycemic improvement, and lipid changes — and no trial has isolated the lipid contribution.9 It would therefore overstate the evidence to say that GLP-1 pathways prevent cardiovascular events by regulating lipids; lipids are a plausible partial contributor within a multifactorial effect.
Vascular-function research adds texture but not certainty. Preclinical studies report that GLP-1 receptor agonists exert anti-atherogenic actions on endothelial cells, monocytes/macrophages, and vascular smooth-muscle cells — reducing foam-cell formation, shifting macrophages toward an anti-inflammatory M2 phenotype, and stabilizing plaque.25 Yet these findings are not uniform: at least one preclinical study found GLP-1 receptor activation reduced hepatic lipid but did not attenuate atherosclerosis progression in diabetic ApoE-deficient mice, and another agent failed to reduce plaque area.26 Such negative and discordant results are a healthy reminder that the vascular story is incomplete.
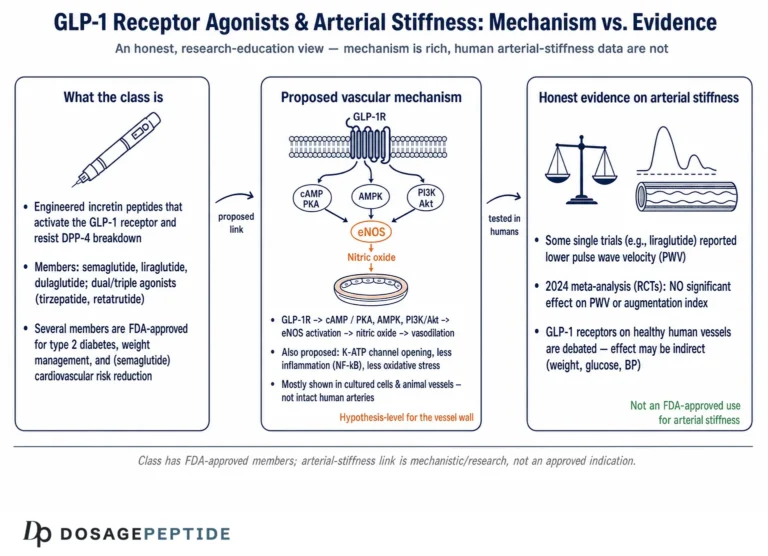
On arterial stiffness specifically — the parameter the title’s framing implicitly invokes — human evidence is genuinely mixed. Some meta-analyses report improvements in flow-mediated dilation and reductions in pulse-wave velocity with GLP-1 receptor agonists, while others conclude the class has no significant effect on classical measures of arterial stiffness.27 A 2025 review of GLP-1 and the endothelium catalogues plausible molecular mechanisms for vascular protection while acknowledging heterogeneous clinical results.28 The fair summary is: endothelial-function signals are encouraging but inconsistent, and a robust, reproducible effect on arterial stiffness has not been established. Anyone asserting that GLP-1 pathways demonstrably reduce arterial stiffness in atherogenic dyslipidemia is currently outrunning the data.
Comparisons: within the class and against lipid-directed therapies
Because “GLP-1 receptor agonist” spans several molecules with different pharmacology, within-class comparison is informative. Shorter-acting exenatide and lixisenatide, which produce pronounced slowing of gastric emptying, tend to show strong postprandial lipid effects; long-acting agents such as semaglutide and dulaglutide produce sustained fasting improvements. Tirzepatide’s added GIP activity appears to confer incrementally larger triglyceride and VLDL reductions than semaglutide in head-to-head data, and investigational triple agonists that add glucagon-receptor activity (e.g., retatrutide) are being studied for even greater hepatic-fat and metabolic effects, though their lipid data are earlier-stage.21 Readers comparing these agents can review the class members directly, for example the tirzepatide dosage protocol and the retatrutide dosage protocol pages, alongside the site’s peptide stacks overview.
A particularly instructive comparison is GIP-receptor agonism as a distinct axis. A 2023 preclinical study reported that GIP-receptor agonism improved dyslipidemia and atherosclerosis independently of body-weight loss in a cardio-metabolic mouse model — a result that helps explain why dual GIP/GLP-1 agonism might exceed GLP-1 alone on lipids, and that supports the broader argument for weight-independent metabolic actions.29 Consistent with this, a post-hoc analysis of the SURMOUNT obesity trials reported that roughly 40% of the changes in VLDL-cholesterol and triglycerides with tirzepatide occurred unassociated with weight reduction.23 These weight-independent fractions are among the most persuasive pieces of evidence that receptor signaling — not just caloric restriction — touches lipid metabolism.
Against dedicated lipid-lowering therapy, the contrast is stark and important for honest framing. Statins lower LDL-cholesterol by 30–55%; ezetimibe and PCSK9 inhibitors add further large LDL reductions; icosapent ethyl and fibrates target triglycerides. GLP-1 receptor agonists produce only single-digit LDL reductions and moderate triglyceride reductions.19 Their appeal in the lipid space is therefore not potency but pleiotropy — simultaneous improvement in weight, glucose, hepatic fat, blood pressure, and triglyceride-rich lipoproteins — and a possible mechanistic reach into the postprandial, remnant-rich biology that statins do not address.4 They complement, rather than replace, lipid-directed drugs where those are indicated.
The comparative bottom line: within the class, agents with stronger gastric-emptying effects or added incretin/glucagon activity tend to show larger lipid effects, and some of that effect is weight-independent. But no GLP-1 receptor agonist rivals purpose-built lipid therapies on the headline LDL metric, and positioning them as lipid drugs would misrepresent both their evidence base and their labeled indications.
Research models and methodology
Understanding how confident to be about any of these claims requires understanding how they are generated. The evidence pyramid for GLP-1 and lipids spans four tiers, each with characteristic strengths and blind spots. At the base are in-vitro systems — HepG2 and primary hepatocytes, enterocyte lines, macrophage cultures — used to probe signaling nodes such as SREBP-2, AMPK, and SIRT6.11 These are mechanistically precise but use supraphysiologic concentrations and cannot capture systemic integration; a pathway demonstrated in a dish is a hypothesis, not a physiological fact.
The second tier is rodent models: diet-induced obese mice, insulin-resistant rats, and atherosclerosis-prone strains such as ApoE-deficient and LDL-receptor-deficient mice. These allow measurement of VLDL production rates, hepatic lipid content, and plaque burden, and they are where much of the direct-versus-indirect debate plays out.8 Their limitation is well known: rodent lipoprotein metabolism differs substantially from human (mice carry cholesterol predominantly in HDL and lack cholesteryl-ester transfer protein), so atherosclerosis models require genetic manipulation and translate imperfectly. The discordant plaque results noted earlier — benefit in some models, none in others — partly reflect these model-dependent quirks.26
The third tier, and the most valuable for the specific question of lipid regulation, is human kinetic and postprandial physiology. Stable-isotope tracer studies measuring apoB48 and apoB100 production and catabolic rates — as in the liraglutide apoB48 study — provide mechanistic resolution in intact humans that neither cell nor rodent work can match, though they are small, intensive, and typically short.13 Meal-challenge and mixed-meal-tolerance-test designs quantify postprandial triglyceride excursions, the parameter most relevant to remnant atherogenicity.15
The fourth tier is large randomized trials and their secondary analyses. These deliver reliable estimates of average lipid change and, in SELECT and the SURPASS/SURMOUNT programs, cardiovascular and body-composition context.24 Their limitation for this question is that lipid endpoints were secondary, confounded by weight loss, and not designed to isolate mechanism. Mediation and weight-adjusted analyses (as in SURMOUNT) partly address this but remain observational within the trial.23 A methodological reader should therefore weight human tracer-kinetic and weight-independent analyses most heavily when judging whether the receptor genuinely “regulates” lipids, and treat single-cell-line mechanistic papers as generative rather than confirmatory.
Safety and tolerability
Because this article concerns an approved class studied in hundreds of thousands of participants, its safety profile is comparatively well characterized — another reason to keep approved and research contexts distinct. The dominant adverse effects are gastrointestinal: nausea, vomiting, diarrhea, and constipation, driven substantially by the same delayed gastric emptying that contributes to postprandial lipid effects.30 These effects are usually dose-dependent, worst during titration, and mitigated by slow escalation, which is why approved products use multi-week up-titration schedules.31
More serious but rarer considerations have been examined extensively. Concerns about acute pancreatitis raised by early pharmacovigilance signals have been substantially tempered by long-term randomized data, though pharmacovigilance databases continue to show agent-level differences, and individual products carry a boxed warning against use in people with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2, based on rodent C-cell tumor findings whose human relevance remains uncertain.31 Additional monitored issues include cholelithiasis (partly linked to rapid weight loss), a possible small increase in heart rate, injection-site reactions, and, in real-world FDA adverse-event analyses, gastrointestinal disorders as the leading reported category.32
The table below organizes the tolerability picture at a class level. It reflects data from approved medical use and is not a research-dosing safety guide; individual agents differ, and definitive safety information resides in the respective regulatory labels.
| Category | Representative effects | Notes on evidence |
|---|---|---|
| Common, usually transient | Nausea, vomiting, diarrhea, constipation | Dose-dependent; worst during titration30 |
| Less common | Cholelithiasis, injection-site reactions, heart-rate increase | Monitored across trials31 |
| Rare / debated | Acute pancreatitis | Early signal largely tempered by RCTs; agent differences persist30 |
| Boxed warning (label) | Thyroid C-cell tumors (rodent finding) | Human relevance uncertain; contraindicated in MEN2/MTC history31 |
Critically for the lipid question, none of this safety profile speaks to using these agents for dyslipidemia, a use for which risk-benefit has not been evaluated in dedicated trials. The safety data support the approved indications (type 2 diabetes, chronic weight management, and specified cardiovascular-risk-reduction uses); extrapolating them to a lipid indication would be unjustified.9
Handling and reconstitution in a research context
Many GLP-1-class research peptides are supplied as lyophilized (freeze-dried) powder that must be reconstituted before laboratory use, and correct handling is a scientific-integrity issue as much as a practical one: degraded or misprepared peptide is a common uncontrolled variable in preclinical work. This section describes handling only as it pertains to research characterization, not human administration.
Lyophilized GLP-1-class peptides are generally reconstituted with bacteriostatic water (sterile water containing 0.9% benzyl alcohol) for multi-use research vials, or plain sterile water for single-use preparations. The volume of diluent is chosen to yield a convenient working concentration; for example, published reconstitution notes for a research semaglutide vial describe adding a defined diluent volume to reach a target concentration for accurate small-volume measurement.33 Diluent should be introduced slowly against the vial wall rather than forced directly onto the powder, and the vial swirled — not shaken — because these peptides are surface-active and vigorous agitation promotes aggregation and foaming that can denature the peptide.
Storage conditions materially affect stability. Lyophilized material is typically stored frozen and protected from light; once reconstituted, aqueous peptide is generally refrigerated at 2–8 °C and used within a limited window (commonly cited as up to about four weeks for bacteriostatic preparations), because peptides in solution are more susceptible to hydrolysis, oxidation, and microbial contamination than the dry powder.33 Repeated freeze-thaw cycles of reconstituted solution should be avoided. Analytical verification — identity and purity by mass spectrometry and HPLC, and confirmation that a batch is what its label claims — is the responsible baseline before any research use, given documented variability in the purity of research-grade peptides from the gray market.
The DosagePeptide.com protocol pages, including the tirzepatide 5 mg protocol and the cagrilintide/semaglutide blend protocol, catalogue reconstitution parameters, concentrations, and storage windows for the specific vial sizes discussed in the literature. These are provided for research reference and are not instructions for human use. It bears repeating that handling proficiency has nothing to do with whether the compound is appropriate or safe for any particular purpose; it only ensures that whatever research is done uses well-characterized material.
Limitations and the human-evidence gap
Having surveyed the mechanisms and data, it is worth consolidating the honest limitations, because they are the heart of why the title is a research question rather than a settled fact. First, indication: no GLP-1 receptor agonist is approved to treat dyslipidemia of any kind. Every lipid figure cited here comes from trials whose primary purpose was glycemic control, weight management, or cardiovascular-event reduction, with lipids as secondary or exploratory measures.20
Second, confounding by weight loss. The single largest driver of lipid improvement in most datasets is the weight reduction these agents produce. Weight-independent analyses (SURMOUNT) and GIP-specific preclinical work suggest a genuine direct component to the lipid effect, but they estimate rather than definitively isolate it, and they apply to specific agents rather than the whole class.23 Until a trial is designed specifically to dissect the lipid mechanism, “regulates lipid metabolism” remains a partially inferential statement.
A further methodological caution concerns generalizability across the class and across populations. Most of the mechanistic and kinetic work has been done with a handful of agents — liraglutide, exenatide, semaglutide, and more recently tirzepatide — and in cohorts selected for type 2 diabetes or obesity. Whether the same lipid biology applies to newer or investigational molecules, to normoglycemic individuals, or to people whose atherogenic dyslipidemia is driven by genetic rather than insulin-resistant causes is largely untested. Extrapolating a favorable postprandial-apoB48 finding from a small diabetic cohort to the broad claim that “the class regulates atherogenic dyslipidemia” stretches the evidence beyond what any single study supports, and it ignores the real pharmacological heterogeneity documented earlier in this article.5
Third, mechanistic uncertainty about receptor location. The debate over whether adult hepatocytes express functional GLP-1 receptors, and the competing evidence for neural and gut-mediated routes, means the field cannot yet draw a clean molecular diagram from receptor to lipoprotein for the fasting hepatic effects.8 The postprandial intestinal axis is better supported, but even there the GLP-1/GLP-2 opposition and the modest contribution of endogenous GLP-1 complicate any simple narrative.18
Fourth, discordant and negative preclinical results. Not every atherosclerosis model shows benefit; some show hepatic-lipid improvement without plaque reduction, and some agents fail to alter plaque at all.26 Fifth, magnitude. Even where effects are real, they are modest for LDL-cholesterol and moderate for triglycerides — not a substitute for dedicated lipid therapy, and of uncertain independent clinical weight given how much larger the concurrent glycemic and weight effects are.19 Finally, arterial-stiffness and endothelial endpoints — the vascular outcomes the title’s phrasing evokes — remain inconsistent across human studies.27 Collectively, these limitations do not diminish the compounds; they define the boundary between what is demonstrated (a favorable lipid-parameter shift, dominant on triglycerides, partly weight-driven) and what is hypothesized (a discrete receptor-driven program that regulates atherogenic dyslipidemia and its vascular consequences).
Regulatory status
Regulatory framing is where the approved-versus-research distinction becomes concrete. Several GLP-1 receptor agonists are fully approved by the U.S. Food and Drug Administration and the European Medicines Agency — for type 2 diabetes (e.g., semaglutide as Ozempic/Rybelsus, dulaglutide, liraglutide, and the dual agonist tirzepatide as Mounjaro), for chronic weight management (semaglutide as Wegovy, tirzepatide as Zepbound), and, in the case of semaglutide 2.4 mg, for reducing the risk of major adverse cardiovascular events in adults with established cardiovascular disease and overweight or obesity following SELECT.24 These are their labeled indications.
None of these approvals is for the treatment of dyslipidemia, atherogenic or otherwise. Lipid effects appear in product labeling and trial reports as observed changes, not as approved therapeutic claims, and regulators have not evaluated these agents against the standard required to market a lipid indication.9 Investigational members of the broader class — including triple agonists such as retatrutide — are in clinical development and are not approved for any indication as of this writing; their lipid and cardiovascular data remain preliminary.2
Separately, the research-peptide market in which many of these compounds circulate as lyophilized powder sits outside the approved-medicine framework entirely. Material sold “for research use only” is not manufactured, tested, or labeled to pharmaceutical standard, is not evaluated by regulators for safety or efficacy in any use, and is not a medicine. The gap between a rigorously regulated prescription product and a research-grade vial of the same nominal peptide is large and consequential, encompassing purity, sterility, dosing accuracy, and clinical oversight. Nothing in this article should be read as endorsing non-approved use of any GLP-1-class compound.
The regulatory summary, then, mirrors the scientific one: the class is real, several members are approved and cardioprotective within defined populations, and the lipid mechanism explored here is an area of legitimate scientific interest that has not risen to the level of an approved therapeutic claim. Treating it as more than that would misstate both the science and its regulatory standing.
Frequently Asked Questions
Do GLP-1 receptor agonists lower cholesterol?
In clinical trials they are associated with modest reductions in LDL-cholesterol and total cholesterol — typically single-digit percentages — and more noticeable reductions in triglycerides.1920 These are far smaller than the LDL reductions achieved by statins or PCSK9 inhibitors. No GLP-1 receptor agonist is approved to treat high cholesterol, and the observed changes are secondary findings from trials designed for glucose, weight, or cardiovascular endpoints, not evidence that these agents should be used as lipid-lowering drugs.
Is atherogenic dyslipidemia an approved reason to use these drugs?
No. Approved indications include type 2 diabetes, chronic weight management, and — for semaglutide 2.4 mg — cardiovascular-risk reduction in specific populations with established cardiovascular disease and overweight or obesity.24 Dyslipidemia is not a labeled indication for any GLP-1 receptor agonist, and using one specifically to treat a lipid disorder would be off-label and unsupported by dedicated outcome trials.
Are the lipid effects just a result of weight loss?
Weight loss is the largest single contributor in most datasets, but not the whole story. Weight-adjusted analyses of tirzepatide (SURMOUNT) estimated that roughly 40% of the triglyceride and VLDL changes occurred independently of weight reduction, and GIP-receptor preclinical work showed dyslipidemia improvement without weight loss.2329 This supports a real weight-independent component, though it has not been fully isolated in a dedicated mechanistic trial.
What is the best-supported mechanism linking GLP-1 to lipids?
The intestinal, postprandial axis has the strongest human evidence. Tracer-kinetic studies show GLP-1 receptor agonists reduce postprandial apoB48 and chylomicron triglyceride by both lowering production and speeding clearance, plausibly via slowed gastric emptying and vagal gut-brain signaling.1316 Hepatic effects on VLDL and cholesterol synthesis are also reported but are more debated, partly because functional hepatocyte GLP-1 receptor expression is uncertain.
Do these drugs reduce arterial stiffness?
The evidence is mixed. Some meta-analyses report improved flow-mediated dilation and reduced pulse-wave velocity, while others find no significant effect on classical arterial-stiffness measures.2728 A reproducible, definitive effect on arterial stiffness has not been established, so claims that GLP-1 receptor agonists reliably reduce arterial stiffness currently outrun the data.
How does tirzepatide compare with semaglutide on lipids?
In head-to-head data (SURPASS-2), tirzepatide produced somewhat larger reductions in triglycerides and VLDL-cholesterol and slightly larger HDL increases than semaglutide, an incremental benefit often attributed to its added GIP-receptor activity.21 The differences are real but modest, and neither agent approaches dedicated lipid therapy on LDL-cholesterol.
Does reducing lipids explain the cardiovascular benefit seen in trials?
Not by itself. The SELECT trial showed a roughly 20% reduction in major adverse cardiovascular events, but participants were already on lipid-lowering therapy, the benefit appeared early, and mediation analyses point to multiple mechanisms — anti-inflammatory, hemodynamic, weight, glycemic, and lipid.24 No trial has isolated the lipid contribution, so it is inaccurate to say the cardiovascular benefit is driven by lipid regulation.
Is research-grade GLP-1 peptide the same as the prescription medicine?
No. Material sold for research use only is not manufactured, tested, or regulated to pharmaceutical standards, may vary in purity and identity, and is not a medicine. It is not evaluated by regulators for safety or efficacy in any use. This distinction matters for purity, sterility, dosing accuracy, and oversight, and nothing here endorses non-approved use of any GLP-1-class compound.
References
- Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metabolism. Review of GLP-1 physiology and pharmacology. PubMed / Cell Press.
- Jastreboff AM, et al. Triple-hormone-receptor agonist retatrutide for obesity — a phase 2 trial. New England Journal of Medicine, 2023. (Investigational GLP-1/GIP/glucagon agonist.)
- Patel VJ, et al. Effect of GLP-1 based therapies on diabetic dyslipidemia. PubMed 24998439. https://pubmed.ncbi.nlm.nih.gov/24998439/
- Ivanova EA, et al. Lipoproteins and Cardiovascular Disease: small, dense LDL and new therapeutic options. PMC8615620. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8615620/
- Nguyen A, et al. GLP (GLP)-1 regulation of lipid and lipoprotein metabolism (review). Medical Review, 2024. https://www.degruyterbrill.com/document/doi/10.1515/mr-2024-0011/html
- Vergès B. Postprandial actions of GLP-1 receptor agonists: the missing link for cardiovascular and kidney protection. Cell Metabolism, 2023. https://www.cell.com/cell-metabolism/fulltext/S1550-4131(23)00004-9
- Hsieh J, et al. GLP1, an important regulator of intestinal lipid metabolism. Arterioscler Thromb Vasc Biol. https://www.ahajournals.org/doi/10.1161/ATVBAHA.115.305479
- Taher J, et al. GLP-1 receptor agonism ameliorates hepatic VLDL overproduction and de novo lipogenesis in insulin resistance. PMC4264039. https://pmc.ncbi.nlm.nih.gov/articles/PMC4264039/
- Marx N, et al. GLP-1 Receptor Agonists for the Reduction of Atherosclerotic Cardiovascular Risk in Patients With Type 2 Diabetes. Circulation. https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.122.059595
- SREBP Regulation of Lipid Metabolism in Liver Disease, and Therapeutic Strategies. PMC10740981. https://pmc.ncbi.nlm.nih.gov/articles/PMC10740981/
- GLP-1 receptor agonists improve cholesterol metabolism by inhibiting SREBP-2 via SIRT6-AMPK pathway in HepG2 cells treated with palmitic acid. J Clin Prev Cardiol / e-jcpp. https://www.e-jcpp.org/journal/view.php?number=109
- Intestinal L-cell mechanoreception regulates hepatic lipid metabolism through GLP-1. Science Advances, 2025. https://www.science.org/doi/10.1126/sciadv.adv3201
- Vergès B, et al. Liraglutide Reduces Postprandial Hyperlipidemia by Increasing ApoB48 Catabolism and Reducing ApoB48 Production in Type 2 Diabetes. Arterioscler Thromb Vasc Biol. https://www.ahajournals.org/doi/10.1161/ATVBAHA.118.310990
- Xiao C, et al. Exenatide, a glucagon-like peptide-1 receptor agonist, acutely inhibits intestinal lipoprotein production in healthy humans. Arterioscler Thromb Vasc Biol, 2012. https://www.ahajournals.org/doi/10.1161/atvbaha.112.246207
- Mather KJ, et al. Improvements in post-challenge lipid response following tirzepatide treatment in type 2 diabetes. Diabetes Obes Metab, 2024. https://dom-pubs.onlinelibrary.wiley.com/doi/full/10.1111/dom.15365
- GLP-1 attenuates intestinal fat absorption and chylomicron production via vagal afferent nerves originating in the portal vein. PMC9486018. https://pmc.ncbi.nlm.nih.gov/articles/PMC9486018/
- Impact of GLP-1 Receptor Agonists on Biochemical Markers of the Initiation of the Atherosclerotic Process. PMC10855444. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10855444/
- Matikainen N, et al. GLP-1 and GLP-2 as Yin and Yang of Intestinal Lipoprotein Production. PMC3554391. https://pmc.ncbi.nlm.nih.gov/articles/PMC3554391/
- Wilding JPH, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity (STEP 1). New England Journal of Medicine, 2021;384:989-1002.
- Impact of semaglutide on lipid profiles in overweight and obese non-diabetic adults: a systematic review and meta-analysis of RCTs. Eur J Pharmacol / ScienceDirect, 2025. https://www.sciencedirect.com/science/article/abs/pii/S0014299925007071
- The Effects of Tirzepatide on Lipid Profile: A Systematic Review and Meta-Analysis of RCTs. PMC11704219. https://pmc.ncbi.nlm.nih.gov/articles/PMC11704219/
- Dahl D, et al. Oral semaglutide improves postprandial glucose and lipid metabolism and delays gastric emptying in type 2 diabetes. PMC8251575. https://pmc.ncbi.nlm.nih.gov/articles/PMC8251575/
- Body-weight-associated and -unassociated changes in lipid profile with tirzepatide: post-hoc analysis of SURMOUNT-1 and SURMOUNT-2. Circulation, 2024 (abstract 4141358). https://www.ahajournals.org/doi/10.1161/circ.150.suppl_1.4141358
- Lincoff AM, et al. Semaglutide and Cardiovascular Outcomes in Obesity Without Diabetes (SELECT). New England Journal of Medicine, 2023;389:2221-2232. ClinicalTrials.gov NCT03574597.
- GLP-1 receptor agonists and atherosclerosis protection: the vascular endothelium takes center stage. Am J Physiol Heart Circ Physiol, 2023. https://journals.physiology.org/doi/full/10.1152/ajpheart.00574.2023
- GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate atherosclerosis in diabetic male ApoE-/- mice. PubMed 23183176. https://pubmed.ncbi.nlm.nih.gov/23183176/
- The effect of SGLT2 inhibitors and GLP-1 receptor agonists on arterial stiffness: a meta-analysis of RCTs. PubMed 38833853. https://pubmed.ncbi.nlm.nih.gov/38833853/
- GLP-1 receptor agonists and the endothelium: molecular and clinical insights into cardiovascular protection. Frontiers in Medicine, 2025. https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1669685/full
- GIP receptor agonism improves dyslipidemia and atherosclerosis independently of body-weight loss in a preclinical mouse model. PMC10436634. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10436634/
- The science of safety: adverse effects of GLP-1 receptor agonists as glucose-lowering and obesity medications. PMC12904723. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12904723/
- Comparative Safety of GLP-1 Receptor Agonists Across Gastrointestinal, Renal and Pancreatic Systems. Pharmaceuticals (MDPI), 2026. https://www.mdpi.com/1424-8247/19/1/136
- Gastrointestinal Safety Assessment of GLP-1 Receptor Agonists in the US: a Real-World FAERS Analysis. PMC11675942. https://pmc.ncbi.nlm.nih.gov/articles/PMC11675942/
- DosagePeptide.com. Semaglutide (10mg Vial) Dosage & Reconstitution Protocol (research reference). https://www.dosagepeptide.com/single-peptide-dosages/semaglutide-10mg-vial-dosage-protocol/
Educational and research-use disclaimer: This article is provided for scientific and educational purposes only. It is not medical advice, and it does not recommend, endorse, or provide instructions for human use of any compound discussed. GLP-1 receptor agonists referenced here include approved prescription medicines with defined, regulator-authorized indications; none is approved for the treatment of dyslipidemia, and the lipid-mechanism topics explored above are areas of ongoing research rather than established therapy. Research-grade peptides sold “for research use only” are not medicines and are not manufactured or evaluated to pharmaceutical standards. Any decisions regarding health, medication, or cardiovascular or lipid disorders should be made only with a qualified, licensed healthcare professional.