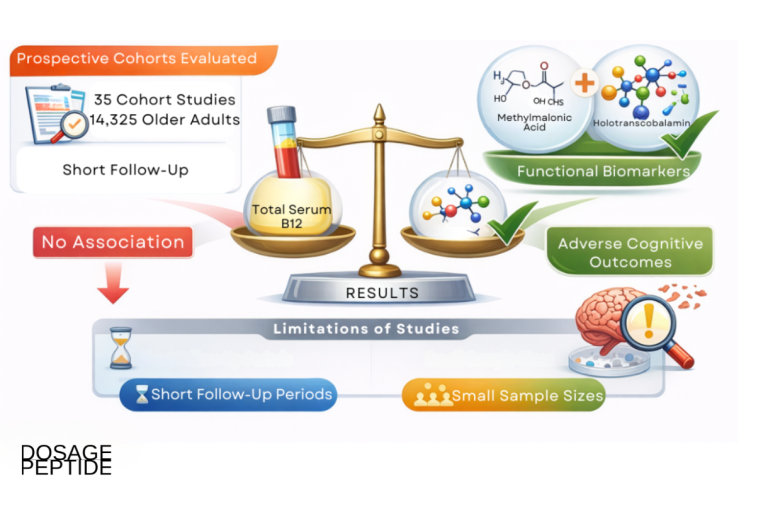
The question in the title is unusually well posed, because for once the biochemistry it presumes is real. Vitamin B12 — cobalamin — genuinely does sit at a control point of cellular methylation. It is the required cofactor for the single enzyme that regenerates methionine from homocysteine, and methionine is the direct precursor of S-adenosylmethionine (SAM), the universal methyl donor that hundreds of methyltransferases draw on to modify DNA, RNA, histones, phospholipids, and small molecules.12 So when someone asks how B12 modulates methylation across cellular research models, they are not chasing a marketing fantasy the way one often is with research peptides. The pathway is textbook, structurally resolved, and reproducible in cell culture, in knockout animals, and in human tissue.
The honest complication lies one layer down, in the word “modulate.” It is easy to slide from “B12 is essential for methylation” to “more B12 means more methylation means better health,” and that slide is where the science stops cooperating. The dependence of methylation on cobalamin is real but it is not linear, and above the threshold needed to keep methionine synthase running, adding cobalamin does not push methylation higher or buy the cell anything measurable. The cellular research models this article surveys make that distinction vivid: remove B12 and methylation collapses in a way you can quantify; restore it and methylation normalizes; flood an already-replete cell and essentially nothing changes. That asymmetry — a steep deficiency effect and a flat repletion effect — is the single most important thing to carry through the whole discussion.
This piece is written for researchers and scientifically literate readers who want an accurate map: what B12 does at the level of atoms and enzymes, how that has been dissected in cellular and animal systems, what deficiency does to the epigenome, and — crucially — where the evidence stops. Along the way it is worth being blunt about a widespread misreading of exactly this biochemistry. Because B12 is a “methylation vitamin,” it is frequently sold as a cure for fatigue, cognitive decline, cardiovascular risk, and aging in people who are not deficient. The large randomized trials that tested that promise did lower homocysteine convincingly and still failed to improve cognition or cardiovascular outcomes in replete populations.789 Holding those two facts together — established mechanism, disappointing supra-repletion outcomes — is the whole discipline of writing honestly about this molecule.
What Vitamin B12 Actually Is
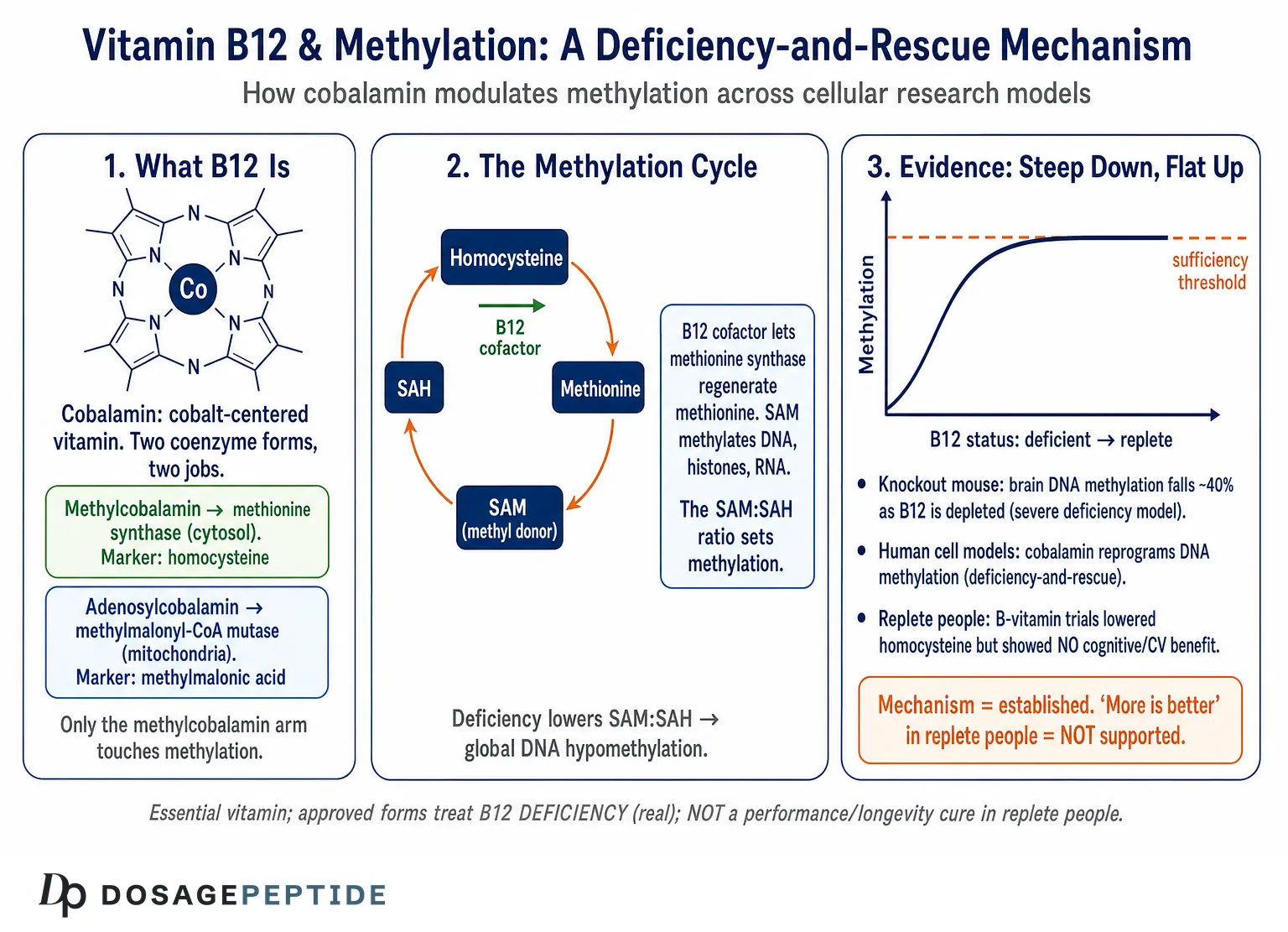
Vitamin B12 is the largest and most structurally elaborate of the vitamins. At its center sits a cobalt ion held in a corrin ring — a macrocycle related to, but distinct from, the porphyrin that cradles iron in heme and magnesium in chlorophyll. Cobalt is what makes cobalamin an organometallic compound, and essentially all of B12’s biological reactivity comes from the chemistry of the bond between that cobalt and whatever group occupies its upper (beta) axial position. Humans cannot synthesize the corrin ring; only certain bacteria and archaea can, which is why dietary B12 ultimately traces back to microbial synthesis and reaches people almost entirely through animal-derived foods.1
The forms of B12 differ only in that upper axial ligand, and the distinction matters enormously for both biochemistry and commerce. Cyanocobalamin carries a cyanide group in that position; it does not occur in appreciable amounts in nature but is the most stable, cheapest, and most common form in supplements and FDA-approved injectables, converted inside the cell to the active coenzymes. Hydroxocobalamin carries a hydroxyl group and is the form used in many parenteral pharmaceutical preparations. The two biologically active coenzyme forms are methylcobalamin (a methyl group on the cobalt) and adenosylcobalamin (a 5′-deoxyadenosyl group). These are not interchangeable trivia: methylcobalamin is the cofactor for methionine synthase in the cytosol, and adenosylcobalamin is the cofactor for methylmalonyl-CoA mutase in the mitochondrion.24 A cell that takes up any B12 form must process it through a dedicated intracellular trafficking pathway that reduces the cobalt and installs the correct upper ligand for each destination.
The reason this molecular preamble matters for a discussion of methylation is that only one of B12’s two jobs — the methylcobalamin-dependent one — touches the methylation cycle directly. When we ask how B12 modulates methylation, we are really asking about a single reaction catalyzed by a single enzyme, and everything downstream flows from whether that reaction is running at capacity. Keeping the two coenzyme forms and their two enzymes cleanly separated is the first habit of clear thinking here, because clinical and biochemical consequences of deficiency split along exactly that line: impaired methionine synthase raises homocysteine and starves methylation, while impaired methylmalonyl-CoA mutase raises methylmalonic acid and disrupts mitochondrial substrate flow.2
| Cobalamin form | Axial group | Role | Compartment / enzyme | Deficiency marker |
|---|---|---|---|---|
| Methylcobalamin | Methyl (–CH₃) | Methyl carrier for remethylation | Cytosol / methionine synthase | Elevated homocysteine |
| Adenosylcobalamin | 5′-deoxyadenosyl | Radical rearrangement cofactor | Mitochondrion / methylmalonyl-CoA mutase | Elevated methylmalonic acid |
| Cyanocobalamin | Cyanide (–CN) | Stable storage/supplement form | Converted intracellularly to active forms | — |
| Hydroxocobalamin | Hydroxyl (–OH) | Common injectable form | Converted intracellularly to active forms | — |
The Central Reaction: Methionine Synthase and the Methyl Group
The reaction at the heart of B12-dependent methylation is deceptively compact. Methionine synthase (also called 5-methyltetrahydrofolate–homocysteine methyltransferase, gene MTR) takes a methyl group from 5-methyltetrahydrofolate and transfers it to homocysteine, producing methionine and regenerating tetrahydrofolate. Cobalamin is the indispensable intermediary: the cobalt shuttles the methyl group in a two-step relay rather than handing it over directly. In the first half-reaction, methylcobalamin donates its methyl group to the sulfur of homocysteine, generating methionine and leaving the enzyme in a highly reactive cob(I)alamin state. In the second half-reaction, that cob(I)alamin is re-methylated by 5-methyltetrahydrofolate, restoring methylcobalamin and releasing tetrahydrofolate.23
Structural work has made this cycle strikingly concrete. Cobalamin-dependent methionine synthase is a large, modular enzyme organized into four functional regions — a homocysteine-binding domain, a 5-methyltetrahydrofolate-binding domain, the cobalamin-binding domain, and a SAM-binding activation domain — and it must undergo dramatic conformational rearrangements to bring the cobalt cofactor alternately into contact with each substrate. A 2023 study captured the full-length enzyme and its cofactor loading in crystallo, resolving how these modules are choreographed so that a single cobalt center can service chemically distinct partners in sequence.3 The mechanistic point that matters for methylation is that the cobalt cycles between the Co(III) methylated state and the Co(I) demethylated state, and that this cycling is what physically links the folate one-carbon pool to the methionine pool.
There is a built-in vulnerability in this design. The cob(I)alamin intermediate is one of the most powerful nucleophiles in biology, and roughly once in every couple of thousand turnovers it is oxidized to an inactive cob(II)alamin state instead of being cleanly re-methylated. When that happens the enzyme stalls, and it can only be rescued by a separate reductive re-methylation that uses SAM as the methyl source, run by the enzyme’s activation domain (with help from methionine synthase reductase). This reactivation requirement is not a footnote; it means methionine synthase is intrinsically prone to going offline and depends on ancillary machinery to keep functioning. It also explains why conditions that oxidize the cobalt — nitrous oxide exposure is the classic example, irreversibly oxidizing cobalamin and inactivating the enzyme — produce a functional methylation deficit even when total B12 stores look adequate.2
Everything the phrase “B12 modulates methylation” means, at the mechanistic level, is contained in this reaction. If methionine synthase is running at capacity, homocysteine is efficiently recaptured into methionine, tetrahydrofolate is regenerated to keep the folate cycle turning, and the methionine pool that feeds SAM synthesis is kept full. If the enzyme is starved of methylcobalamin, homocysteine accumulates, folate gets trapped, and SAM production falters — the three biochemical signatures of impaired methylation that recur across every research model discussed below.
From Methionine to SAM: The Universal Methyl Donor
Methionine is not itself the methyl donor for cellular methylation; it is the raw material. Methionine adenosyltransferase condenses methionine with ATP to form S-adenosylmethionine, and it is SAM that virtually every methyltransferase in the cell uses as its methyl source. When a methyltransferase transfers SAM’s activated methyl group to a substrate — a cytosine in DNA, a lysine or arginine in a histone tail, a nitrogen in a neurotransmitter, a head group in a phospholipid — the product is S-adenosylhomocysteine (SAH). SAH is then hydrolyzed to homocysteine, and homocysteine stands at a fork: it can be remethylated back to methionine (the B12-dependent route that closes the loop) or diverted irreversibly into the transsulfuration pathway toward cysteine and glutathione.2
The ratio of SAM to SAH is often called the cell’s methylation potential, and it is the variable that actually governs methyltransferase activity. This is a subtle and frequently missed point. SAH is not merely a spent byproduct; it is a potent product-inhibitor of most methyltransferases. So methylation is throttled not simply by how much SAM is present but by the SAM:SAH ratio, and anything that lets SAH accumulate — including a homocysteine backup caused by failing remethylation — suppresses methyltransferase activity from the product side. When B12 deficiency stalls methionine synthase, homocysteine rises, the equilibrium of the SAH hydrolase reaction is pushed backward, SAH climbs, and the SAM:SAH ratio falls. Methylation is inhibited by both a fall in the donor and a rise in the inhibitor. This dual-hit dynamic is why cobalamin status can have a disproportionate effect on the epigenome relative to how small the enzymatic lesion looks in isolation.2
It is worth pausing here to see why the “more is better” intuition fails at exactly this junction. Once methionine synthase is saturated with cofactor and remethylation is keeping pace with SAH hydrolysis, the SAM:SAH ratio is set by the fluxes through the cycle, not by the availability of cobalamin. Adding cobalamin to a cell that already has enough does not raise SAM, does not lower SAH, and does not increase global methylation — the rate-limiting steps have moved elsewhere (methionine intake, folate status, the demand from methyltransferases). The vitamin behaves like a key that either fits the lock or does not; a drawer full of spare keys does not open the door any wider. That is the biochemical basis for the clinical observation, developed later, that supplementing replete people yields no methylation-dependent benefit.
The Folate Interlock and the Methyl Trap
B12-dependent methylation cannot be understood in isolation from folate, because methionine synthase is the one enzyme that both cycles share, and it is the hinge on which they turn. The folate cycle carries one-carbon units in various oxidation states for purposes ranging from purine and thymidylate synthesis to remethylation. The specific species that feeds methionine synthase is 5-methyltetrahydrofolate, and here lies one of the most instructive phenomena in all of one-carbon metabolism: the methyl folate trap.2
The reaction that produces 5-methyltetrahydrofolate, catalyzed by methylenetetrahydrofolate reductase (MTHFR), is physiologically irreversible. Once a folate molecule is reduced to the 5-methyl form, the only biologically significant way to get it back into the general tetrahydrofolate pool is for methionine synthase to strip that methyl group off and hand it to cobalamin. If methionine synthase is not working — because cobalamin is deficient — then folate accumulates in the 5-methyl form and is functionally trapped, unable to participate in the other one-carbon reactions the cell needs. The paradoxical result is that B12 deficiency produces a functional folate deficiency even when folate intake and total folate levels are normal. This is the mechanistic explanation for one of the oldest observations in hematology: the megaloblastic anemia of B12 deficiency is indistinguishable at the level of the bone marrow from that of folate deficiency, because in both cases the shared downstream lesion is impaired thymidylate synthesis and stalled DNA replication.12
The methyl trap has a clinically important corollary that bears directly on how methylation is studied and managed. Giving folic acid to a B12-deficient person can partially correct the anemia — by supplying reduced folate through a route that bypasses the trap — while doing nothing for the methylation deficit or the neurological damage, and potentially masking the deficiency until irreversible nerve injury has occurred. This is precisely why B12 status must be evaluated independently and why mandatory folic acid fortification programs are designed with B12 masking in mind. For the methylation question specifically, the trap illustrates that cobalamin and folate are not redundant methyl sources; they are serial links in one chain, and the cobalamin link is the one that closes the methionine loop. Readers who want the underlying vocabulary — remethylation, transsulfuration, one-carbon units — laid out term by term may find the site’s research glossary a useful companion.
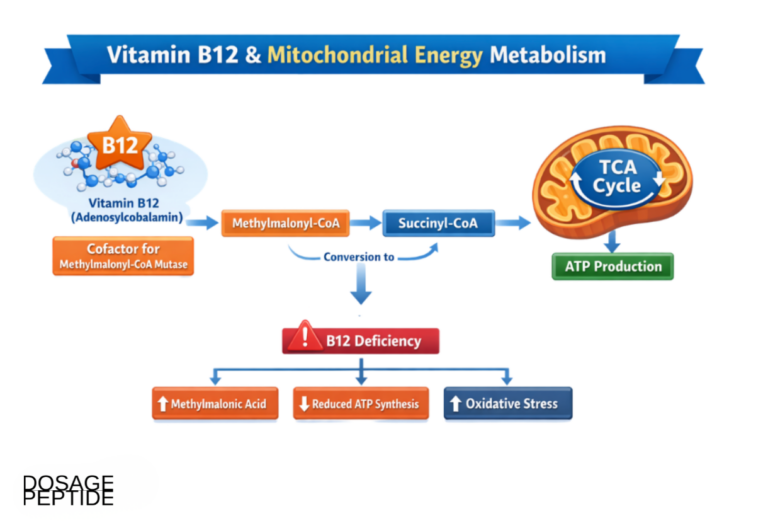
The Second Job: Adenosylcobalamin and Mitochondrial Metabolism
Although this article is about methylation, the second B12-dependent reaction deserves a clear description, both because it is the source of the other major deficiency marker and because it clarifies what B12 does not do in the methylation cycle. In the mitochondrion, adenosylcobalamin serves as the cofactor for methylmalonyl-CoA mutase, which catalyzes the isomerization of L-methylmalonyl-CoA to succinyl-CoA — a step in the catabolism of odd-chain fatty acids, branched-chain amino acids, and cholesterol side chains that funnels these substrates into the citric acid cycle.4
The chemistry here is entirely different from the methyl-transfer of methionine synthase. Rather than shuttling a methyl group, adenosylcobalamin works by homolysis of the cobalt–carbon bond to generate a 5′-deoxyadenosyl radical, which abstracts a hydrogen atom from the substrate and initiates a radical-based carbon-skeleton rearrangement. It is a radical isomerase, not a methyltransferase.4 This matters for the present discussion in a specific way: the mitochondrial reaction has no direct role in DNA, RNA, or histone methylation. When methylmalonyl-CoA mutase fails from B12 deficiency, methylmalonic acid accumulates and can be measured in blood and urine as a sensitive functional marker of cobalamin status, but the epigenetic consequences of deficiency run almost entirely through the cytosolic, methionine-synthase arm.2
Keeping the two arms distinct prevents a common conflation. Because methylmalonic acid is the more specific marker of B12 status — homocysteine also rises in folate and B6 deficiency, whereas methylmalonic acid is comparatively selective for cobalamin — it is tempting to treat the mitochondrial reaction as the “main” B12 job. For methylation questions, the opposite is true: the mitochondrial reaction is the better diagnostic flag, but the cytosolic reaction is the one doing the methylation biology. A researcher using methylmalonic acid to confirm that a cell model is genuinely B12-depleted is leveraging one arm to certify the state of the other.
How Methylation Is Studied Across Cellular Research Models
The title’s phrase — “across cellular research models” — points at the real strength of the B12/methylation field: because the pathway is defined enzyme by enzyme, it can be perturbed and read out in systems ranging from purified protein to whole organism. Understanding what each model can and cannot show is essential to reading the literature honestly.
Purified enzyme and structural systems. At the most reductionist level, methionine synthase has been expressed, crystallized, and interrogated with the cobalt cofactor in defined states, allowing direct observation of the methyl-transfer geometry and the conformational gymnastics required to serve multiple substrates.3 These systems answer the “how does the chemistry work” question with atomic precision but say nothing about physiology; they show mechanism, not consequence.
Cultured mammalian cells. The workhorse for connecting cobalamin status to the epigenome is cell culture, where B12 in the medium can be manipulated and global or gene-specific methylation read out. A representative example is a study of human ileal epithelial cells in which vitamin B12 availability was shown to reprogram transcriptional, metabolic, and epigenetic states, including DNA-methylation changes — direct evidence that cobalamin supply, acting through the SAM-generating pathway, shapes the methylation landscape of a human cell.5 Cell models are powerful because they isolate the cobalamin variable, but they carry an important caveat for the honesty of any claim: culture systems are frequently run at cobalamin concentrations far from physiological, and an effect seen when cells are moved from near-zero B12 to abundant B12 demonstrates the deficiency response, not a benefit of supra-physiological loading.
Genetic knockout and receptor models. Deleting components of the cobalamin uptake or trafficking machinery lets researchers create clean, tissue-specific B12 deprivation in a living animal. The transcobalamin-receptor (TCblR/CD320) knockout mouse is instructive: brain cobalamin falls by as much as 90% over twenty weeks, and this is accompanied by roughly a 40% drop in global DNA methylation in the brain — a striking, quantifiable demonstration that impairing cellular B12 uptake starves the methylation cycle and hypomethylates the genome.6 Knockout models establish causality in vivo in a way culture cannot, but the manipulation is severe by design and models profound deficiency rather than the marginal status seen in most people.
Human observational and interventional data. At the top of the hierarchy sit human studies — measurements of methylation status against cobalamin status, and randomized trials of supplementation. These are the most relevant to real-world claims and, as detailed below, also the most sobering, because they repeatedly show that the steep effects seen in deficiency models do not translate into benefits when B12-replete people are given more.789
| Model tier | What it manipulates | What it can show | Key limitation |
|---|---|---|---|
| Purified enzyme / structure | Cofactor state, protein conformation | Catalytic mechanism of methyl transfer3 | No physiological consequence |
| Cultured mammalian cells | B12 concentration in medium | Epigenetic/transcriptional reprogramming by cobalamin5 | Often non-physiological B12 ranges |
| Genetic knockout animals | Uptake/trafficking genes (e.g. CD320) | Causal link: deficiency → genomic hypomethylation6 | Models severe, not marginal, deficiency |
| Human observation / RCT | Dietary or supplemental B12 | Real-world methylation and clinical outcomes78 | Repletion rarely adds benefit above threshold |
The methodological throughline is that the models agree on the shape of the dependence. Each tier confirms that removing cobalamin impairs methylation and restoring it repairs methylation, and none of them shows that pushing cobalamin above sufficiency increases methylation further. That convergence is what licenses a confident mechanistic statement and, simultaneously, a cautious clinical one.
What Deficiency Reveals: DNA Methylation and Epigenetics
The epigenetic reach of cobalamin is broad because SAM is the common currency of essentially all biological methylation. When B12 deficiency lowers the SAM:SAH ratio, the effect is not confined to one class of substrate; DNA methyltransferases, histone methyltransferases, and RNA and small-molecule methyltransferases all draw on the same donor pool and all feel the same product-inhibition from rising SAH. In principle, then, cobalamin status can influence the entire methyl-dependent regulatory apparatus of the cell.2
The most heavily studied readout is DNA methylation, because global 5-methylcytosine content and gene-specific promoter methylation are technically accessible and biologically consequential. The direction of effect in deficiency is consistent: hypomethylation. The CD320 knockout brain loses roughly 40% of its global DNA methylation as cobalamin is depleted,6 and human ileal epithelial cell work shows that cobalamin availability reshapes the DNA-methylation and transcriptional program.5 Because DNA methylation participates in transposon silencing, imprinting, X-inactivation, and tissue-specific gene regulation, global hypomethylation is not a benign cosmetic change; it is associated with genomic instability and aberrant gene expression in the models where it has been probed.
There is a developmental dimension that raises the stakes further. One-carbon metabolism is under intense demand during rapid cell division and during the epigenetic reprogramming of early development, which is why maternal folate and B12 status has been linked to methylation patterns in offspring in animal models, and why B12 deficiency is one of the nutritional factors implicated in the neural-tube-defect literature alongside folate.1 This is a legitimate and important area — but it is also one where honest framing requires care. The strong evidence concerns deficiency and correcting deficiency (the established public-health case for adequate periconceptional folate, with B12 as a co-factor in the same cycle); it is not evidence that supra-adequate cobalamin optimizes the epigenome of a well-nourished individual.
The intellectually honest summary of the epigenetic section is therefore two-sided. On one hand, the claim that B12 modulates methylation is not merely plausible but demonstrated across model systems, with a clear and consistent mechanism and a reproducible direction of effect. On the other hand, every one of these demonstrations is a deficiency-and-rescue result. The epigenome responds to the presence or absence of adequate cobalamin, not to abundance beyond adequacy. For a broader view of how methyl-donor and cofactor availability intersects with DNA integrity and repair — a related but distinct branch of the same nutritional-biochemistry tree — the discussion of NAD+ and DNA repair mechanisms offers a useful contrast in how another essential cofactor is studied with the same deficiency-versus-repletion logic.
The Replete-Versus-Deficient Distinction: Where the Premise Breaks Down
This is the section the whole article has been building toward, because it is where a real mechanism collides with an overstated promise. If B12 raises the methylation potential and methylation governs gene expression, neuronal maintenance, and vascular biology, then it is tempting to predict that giving B12 — or the broader homocysteine-lowering B-vitamin combination — should improve cognition, slow aging, and reduce cardiovascular events. That hypothesis was not left to speculation. It was tested in large, well-designed randomized controlled trials, and the results are the clearest available reality check on the “methylation cure” narrative.
Cognition. In a two-year, double-blind, placebo-controlled trial of 276 healthy older adults with elevated homocysteine, daily folate plus B12 and B6 lowered plasma homocysteine by an impressive 4.36 µmol/L relative to placebo — and produced no significant improvement on any test of cognition.7 That single trial might be dismissed as underpowered, but it is not an outlier. A meta-analysis pooling 11 trials with cognitive data on roughly 22,000 individuals found that B-vitamin supplementation lowered homocysteine by around a quarter to a third and had no significant effect on global cognitive function or on individual cognitive domains.8 The biochemistry worked exactly as designed — homocysteine fell, implying the methylation cycle was being pushed — and the clinical payoff in these largely replete populations was absent.
Cardiovascular outcomes. The same pattern holds for the vascular hypothesis that dominated homocysteine research for a decade. The HOPE-2 trial randomized 5,522 patients with vascular disease or diabetes to folic acid plus B12 and B6 or placebo; the intervention lowered homocysteine but did not reduce the primary composite of cardiovascular death, myocardial infarction, and stroke.9 Across the broader trial literature, homocysteine-lowering with B vitamins has not delivered consistent reductions in cardiovascular events, and homocysteine has accordingly been reframed by much of the field as a marker of risk rather than a validated causal target — the classic epidemiological trap where lowering a biomarker fails to move the outcome it flagged.
The honest exception. Scientific integrity requires naming the one recurring positive signal. In the VITACOG trial, B-vitamin supplementation slowed the rate of brain atrophy in older adults with mild cognitive impairment, with the benefit concentrated in those with higher baseline homocysteine.10 This is a genuine, peer-reviewed finding, and it points to the correct interpretation of the entire body of trial data: benefit appears where there is a deficit to correct (elevated homocysteine, likely reflecting marginal B-vitamin status), and disappears where status is already adequate. It is a deficiency-correction result, not evidence that B12 is a cognitive enhancer for the replete. Notably, even VITACOG measured a surrogate (atrophy rate) rather than proven prevention of dementia, and attempts to generalize it into a broad “B12 protects the aging brain” claim run well ahead of the evidence.
| Trial / analysis | Population | Homocysteine effect | Clinical outcome |
|---|---|---|---|
| McMahon 2006 (RCT)7 | 276 healthy older adults, high Hcy | Lowered by 4.36 µmol/L | No cognitive improvement |
| Clarke 2014 (meta-analysis)8 | ~22,000 individuals, 11 trials | Lowered ~26–28% | No effect on cognition |
| HOPE-2 2006 (RCT)9 | 5,522 vascular/diabetes patients | Lowered | No reduction in CV death/MI/stroke |
| VITACOG 2010 (RCT)10 | Older adults with MCI, elevated Hcy | Lowered | Slowed brain atrophy (surrogate) |
The lesson these trials teach about methylation specifically is precise and worth stating without hedging: lowering homocysteine is not the same as improving health. The B vitamins reliably moved the methylation-cycle biomarker, and that biochemical success did not translate into the clinical benefits the mechanism seemed to promise. Anyone reasoning from “B12 modulates methylation” to “B12 supplementation improves outcomes” is making a leap the randomized evidence has repeatedly refused to support in replete populations. The mechanism is real; the extrapolation is not licensed. This is the same discipline of separating a demonstrated pathway from an unproven clinical payoff that governs honest writing about age-related interventions generally, a theme explored in the analysis of whether growth-hormone secretagogues support cognition in aging.
Where the Mechanism Does Deliver: Genuine B12 Deficiency
It would be a distortion of the opposite kind to leave the impression that B12 does nothing. In the specific, definable state of deficiency, cobalamin is not a marginal-benefit supplement but an essential, sometimes life-saving, treatment — and this is one of the clearest examples in medicine of correcting a methylation defect at its source. Clinical B12 deficiency, whether from dietary inadequacy (strict plant-based diets without supplementation), malabsorption (atrophic gastritis, the autoimmune loss of intrinsic factor that defines pernicious anemia, gastric or ileal surgery, or long-term acid suppression and metformin use), produces two well-characterized syndromes that map onto the two coenzyme functions.1
The hematological syndrome is megaloblastic anemia, driven by the methyl-folate trap’s downstream impairment of DNA synthesis in erythroid precursors. The neurological syndrome — subacute combined degeneration of the spinal cord, peripheral neuropathy, and cognitive changes — is thought to reflect, at least in part, the methylation failure affecting myelin maintenance and central nervous system biochemistry.1 The critical clinical fact is that the neurological damage can become irreversible if deficiency is prolonged, which is why prompt and adequate repletion matters and why folic acid must never be used to paper over an unrecognized B12 deficiency. Parenteral cyanocobalamin or hydroxocobalamin corrects the hematological abnormalities reliably and halts neurological progression, and patients with pernicious anemia require lifelong replacement because the underlying malabsorption does not resolve.1112
This is the honest home of the methylation mechanism. In deficiency, restoring cobalamin restores methionine synthase activity, refills the SAM pool, relieves the SAH-mediated inhibition of methyltransferases, releases the folate trap, and reverses the hematological consequences — a chain of causation that runs directly through the methylation cycle this article has described. The demonstrated power of B12 is a power to correct a defined deficiency state, not to enhance a normal one.
B12 in a Research and Dosing Context
Because vitamin B12 is encountered both as an approved medicine and, increasingly, as an injectable compound circulating in research and wellness settings, a brief, strictly educational note on forms and handling is warranted — with the emphasis that established medical use of B12 is for the treatment of deficiency, not for methylation “optimization” in replete individuals.
The dietary reference intake for adults is modest — on the order of 2.4 micrograms per day — a quantity that underlines how little cobalamin the body actually requires to keep its two enzymes supplied, and how the vitamin’s efficient enterohepatic recycling and hepatic storage buffer intake over long periods.13 This is the arithmetic behind one of the most important framing points in the whole subject: the gap between the microgram-scale physiological requirement and the milligram-scale doses common in injectable products is enormous, and the excess is not doing proportional extra methylation work. Water-soluble cobalamin above the saturation of its transport and enzymatic capacity is largely excreted, which is also why acute toxicity is low but why high-dose regimens should not be mistaken for high-dose benefit.
In a laboratory or compounding context, injectable B12 is most often supplied as cyanocobalamin or hydroxocobalamin in solution, or as a lyophilized powder for reconstitution. The general principles that apply to any injectable biologic apply here: cobalamin is notably light-sensitive (its solutions are the characteristic deep red, and photodegradation is real), so vials are protected from light; solutions are kept cool; and aseptic technique governs any reconstitution. The concentration arithmetic is the same as for any reconstituted compound — a fixed mass of cobalamin dissolved in a chosen volume of diluent sets the concentration per unit volume — and general walk-throughs of that arithmetic appear on the site’s reconstitution guide and dosage calculator, which are provided for educational reference rather than as instructions for human use. It bears repeating that meticulous handling changes nothing about the underlying evidence: a perfectly prepared high-dose vial administered to a replete person still faces the flat repletion curve that the clinical trials documented.
A researcher cataloging cobalamin alongside other compounds by form and handling parameters will find it grouped, on the site’s central dosage index, among water-soluble injectables — a classification that reflects its chemistry, not any endorsement of the methylation-optimization claims that often accompany its marketing.
Safety and Tolerability
Vitamin B12 has one of the widest safety margins of any bioactive compound, a fact that is genuine and that is also, paradoxically, part of why overstated claims proliferate around it — a molecule that rarely causes harm is an easy one to recommend indiscriminately. Because cobalamin is water-soluble and excess is efficiently excreted, no tolerable upper intake level has been established on the basis of toxicity, and high oral or parenteral doses are generally well tolerated.1113 Reported adverse effects of injectable cyanocobalamin are usually mild and infrequent, and serious reactions are rare.12
Several caveats keep this reassuring picture honest:
- Low toxicity is not evidence of benefit. The most common error is to reason that because B12 is safe, taking more “can’t hurt and might help.” The trial data show that in replete people it reliably does not help with the outcomes it is marketed for, and safety says nothing about efficacy.
- Masking of deficiency. The interaction that matters most is between B12 and folic acid: high folic acid intake can correct the anemia of B12 deficiency while allowing neurological damage to progress undetected. This is a reason for caution in indiscriminate high-dose B-vitamin use, not a property of B12 itself.1
- Rare hypersensitivity. Allergic and, rarely, anaphylactoid reactions to cobalamin injections have been reported; a documented sensitivity is a contraindication.12
- Product quality outside regulated channels. Injectable B12 sold as a “research” or wellness product outside pharmacy dispensing may vary in purity, sterility, and labeling accuracy — risks that attach to sourcing rather than to the molecule.
- Confounding in high-risk populations. Some observational data have linked high circulating B12 to adverse outcomes in certain ill populations, but elevated serum B12 in those settings is generally a consequence of underlying disease (releasing bound cobalamin) rather than a cause of harm — a reminder that association is not causation in either direction.
The balanced reading is that cobalamin is safe enough that safety is rarely the limiting consideration; the limiting consideration is whether there is anything to correct. Absence of harm and absence of benefit coexist comfortably in the replete individual.
Regulatory Status
The regulatory picture for vitamin B12 is unusually clean compared with most compounds discussed in a research-peptide context, and precision about what is and is not approved prevents the mechanism from being oversold.
Approved for deficiency. Cyanocobalamin (and hydroxocobalamin) injection is an FDA-approved drug for the treatment of vitamin B12 deficiency, including pernicious anemia and deficiency arising from malabsorption, gastrointestinal surgery, and dietary inadequacy.1112 This is a real, long-standing, evidence-based approval, and it is the correct anchor for any statement about what B12 is proven to do: it treats and prevents the consequences of cobalamin deficiency. The approval is specific to correcting the deficiency state, not to enhancing methylation, cognition, energy, or longevity in people whose B12 status is normal.
Dietary-supplement framing. Beyond the approved injectable indications, the vast majority of B12 consumed is sold as a dietary supplement (oral tablets, sublingual products, and lozenges). Under the U.S. dietary-supplement framework, these products may not lawfully claim to treat, cure, or prevent disease, and their marketing operates under structure/function language. It is precisely within this framework that the “methylation support,” “energy,” and “brain health” positioning flourishes — claims that are not evaluated as drug-efficacy claims and that, for replete consumers, are not supported by the interventional evidence reviewed above.
The nutritional-requirement basis. B12’s status as an essential nutrient with an established dietary reference intake is a matter of settled nutritional science, and adequate intake is a legitimate public-health goal, especially for populations at risk of deficiency: older adults with reduced absorption, people following strict plant-based diets, and those on long-term metformin or acid-suppressing therapy.113 Recommending adequate B12 to prevent deficiency is sound; implying that supplementation optimizes the methylation-dependent health of the already-adequate is where the science is left behind.
The regulatory synthesis is straightforward and, unlike many compounds, not ambiguous: B12 is an essential vitamin, its deficiency states are real and its deficiency treatment is FDA-approved, and its methylation biochemistry is textbook — but there is no regulatory recognition, and no supporting trial evidence, for using it as a methylation enhancer or performance/longevity agent in replete people.
Frequently Asked Questions
Does vitamin B12 really affect DNA methylation?
Yes — this part of the premise is genuinely well established. B12 (as methylcobalamin) is the required cofactor for methionine synthase, the enzyme that regenerates methionine from homocysteine. Methionine is converted to S-adenosylmethionine (SAM), the methyl donor for DNA methyltransferases and essentially all other cellular methylation.2 When cobalamin is deficient, the SAM:SAH ratio falls, methyltransferases are inhibited, and global DNA methylation drops — shown directly in knockout mice (roughly a 40% fall in brain DNA methylation) and in human cell models.56 The important nuance is that this is a deficiency effect; adding B12 to an already-replete cell does not push methylation higher.
Will taking more B12 boost my methylation or energy if I’m not deficient?
The evidence says no. B12 acts like a cofactor that either fits its enzyme or does not; once methionine synthase is supplied, extra cobalamin does not raise SAM or increase methylation, and the excess is largely excreted. Large randomized trials that lowered homocysteine (a direct methylation-cycle marker) with B vitamins found no improvement in cognition in generally replete older adults.78 Low toxicity means high doses rarely harm, but “won’t hurt” is not the same as “will help.”
Why did B12 fail in the cognitive and cardiovascular trials if the mechanism is real?
Because the trials mostly enrolled people who were not meaningfully deficient, and the mechanism only has room to work when there is a deficit to correct. Homocysteine-lowering with B vitamins reliably moved the biomarker but did not reduce cardiovascular events (HOPE-2) or improve cognition (multiple trials and a 22,000-person meta-analysis).89 Homocysteine turned out to be a marker of risk rather than a causal target you can simply lower for benefit — a classic case where fixing a biomarker does not fix the outcome.
Is there any situation where B12 clearly helps the brain?
Yes, but a narrow one. In the VITACOG trial, B vitamins slowed brain atrophy in older adults with mild cognitive impairment and elevated homocysteine — the benefit concentrated in those with the highest baseline homocysteine, i.e. those most likely to have a correctable deficit.10 This is a deficiency-correction signal on a surrogate endpoint (atrophy rate), not proof that B12 prevents dementia or enhances the healthy brain.
What is the methyl-folate trap, and why does it matter?
When B12 is deficient, methionine synthase stalls, and folate becomes stuck in its 5-methyl form because the reaction that made it is irreversible and only methionine synthase can free it.2 This creates a functional folate deficiency even with normal folate intake — which is why B12 and folate deficiency cause an identical megaloblastic anemia, and why giving folic acid can mask B12 deficiency while nerve damage silently progresses.1
How is B12’s effect on methylation actually studied?
Across a hierarchy of models: purified enzyme and crystal structures show the methyl-transfer chemistry;3 cultured human cells show cobalamin reprogramming DNA methylation and transcription;5 genetic knockout animals show that cutting off cellular B12 uptake causes genomic hypomethylation;6 and human trials test whether supplementation changes clinical outcomes.78 Each tier converges on the same shape: deficiency impairs methylation, repletion restores it, and excess adds nothing.
What are the two different jobs of B12?
Methylcobalamin is the cofactor for methionine synthase in the cytosol (the methylation-cycle enzyme, deficiency marker: high homocysteine), and adenosylcobalamin is the cofactor for methylmalonyl-CoA mutase in the mitochondrion (a radical isomerase in energy metabolism, deficiency marker: high methylmalonic acid).24 Only the first touches methylation directly; the second is the more specific diagnostic flag for deficiency.
Is B12 approved by the FDA?
Cyanocobalamin and hydroxocobalamin injections are FDA-approved for treating vitamin B12 deficiency, including pernicious anemia and malabsorption.1112 That approval is specifically for correcting deficiency. Oral B12 supplements are sold under the dietary-supplement framework and may not lawfully claim to treat disease; there is no approval for using B12 as a methylation enhancer or performance agent in people who are not deficient.
Who is actually at risk of B12 deficiency?
Older adults (reduced stomach acid and intrinsic factor), people on strict plant-based diets without supplementation, those with pernicious anemia or gastrointestinal surgery, and long-term users of metformin or acid-suppressing drugs.113 For these groups, ensuring adequate B12 is genuinely important and is where the mechanism delivers real benefit — by correcting a deficiency, not by optimizing an already-normal state.
References
- Green R, Allen LH, Bjørke-Monsen AL, et al. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017;3:17040. PMID 28660890. https://www.nature.com/articles/nrdp201740
- Froese DS, Fowler B, Baumgartner MR. Vitamin B12, folate, and the methionine remethylation cycle—biochemistry, pathways, and regulation. J Inherit Metab Dis. 2019;42(4):673-685. PMID 30693532. https://pubmed.ncbi.nlm.nih.gov/30693532/
- Mendoza J, Purchal M, Yamada K, Koutmos M. Structure of full-length cobalamin-dependent methionine synthase and cofactor loading captured in crystallo. Nat Commun. 2023;14:6365. PMID 37821448. PMC10567725. https://www.nature.com/articles/s41467-023-42037-4
- Takahashi-Iñiguez T, García-Hernandez E, Arreguín-Espinosa R, Flores ME. Role of vitamin B12 on methylmalonyl-CoA mutase activity. J Zhejiang Univ Sci B. 2012;13(6):423-437. PMID 22661206. PMC3370288. https://pmc.ncbi.nlm.nih.gov/articles/PMC3370288/
- Ge Y, Zadeh M, Mohamadzadeh M. Vitamin B12 Regulates the Transcriptional, Metabolic, and Epigenetic Programing in Human Ileal Epithelial Cells. Nutrients. 2022;14(14):2825. PMID 35889782. PMC9321803. https://pmc.ncbi.nlm.nih.gov/articles/PMC9321803/
- Fernàndez-Roig S, Lai SC, Murphy MM, Fernandez-Ballart J, Quadros EV. Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr Metab (Lond). 2012;9:41. PMID 22553938. PMC3433370. https://pmc.ncbi.nlm.nih.gov/articles/PMC3433370/
- McMahon JA, Green TJ, Skeaff CM, Knight RG, Mann JI, Williams SM. A controlled trial of homocysteine lowering and cognitive performance. N Engl J Med. 2006;354(26):2764-2772. PMID 16807413. https://www.nejm.org/doi/full/10.1056/NEJMoa054025
- Clarke R, Bennett D, Parish S, et al. Effects of homocysteine lowering with B vitamins on cognitive aging: meta-analysis of 11 trials with cognitive data on 22,000 individuals. Am J Clin Nutr. 2014;100(2):657-666. PMID 24965307. https://pmc.ncbi.nlm.nih.gov/articles/PMC4095663/
- Lonn E, Yusuf S, Arnold MJ, et al (Heart Outcomes Prevention Evaluation 2 Investigators). Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006;354(15):1567-1577. PMID 16531613. https://www.nejm.org/doi/full/10.1056/NEJMoa060900
- Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5(9):e12244. PMID 20838622. PMC2935890. https://pmc.ncbi.nlm.nih.gov/articles/PMC2935890/
- Ankar A, Kumar A. Vitamin B12 Deficiency. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024. NBK441923. https://www.ncbi.nlm.nih.gov/books/NBK441923/
- U.S. Food and Drug Administration. Cyanocobalamin Injection, USP 1000 mcg/mL — prescribing information (ANDA 080737). https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/080737Orig1s040lbl.pdf
- National Institutes of Health, Office of Dietary Supplements. Vitamin B12: Fact Sheet for Health Professionals. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
Educational and research-use disclaimer: This article is provided solely for scientific and educational purposes. Vitamin B12 (cobalamin) is an essential nutrient, and FDA-approved cyanocobalamin and hydroxocobalamin injections are indicated for the treatment of vitamin B12 deficiency, including pernicious anemia. Its role in the methylation cycle is well established. However, B12 is not approved, and current randomized evidence does not support its use, as a methylation enhancer, cognitive enhancer, cardiovascular preventive, or longevity or performance agent in individuals who are not deficient. Nothing here is medical advice or a recommendation for self-administration. B12 status should be assessed and deficiency managed by qualified healthcare professionals, and any injectable use should occur through appropriate medical or licensed channels. Readers should consult qualified professionals and applicable regulations before making any decisions.